Judit Burgaya, Julie Marin, Guilhem Royer, Bénédicte Condamine, Benoit Gachet, Olivier Clermont, Françoise Jaureguy, Charles Burdet, Agnès Lefort, Victoire de Lastours, Erick Denamur, Marco Galardini, François Blanquart
{"title":"The bacterial genetic determinants of Escherichia coli capacity to cause bloodstream infections in humans.","authors":"Judit Burgaya, Julie Marin, Guilhem Royer, Bénédicte Condamine, Benoit Gachet, Olivier Clermont, Françoise Jaureguy, Charles Burdet, Agnès Lefort, Victoire de Lastours, Erick Denamur, Marco Galardini, François Blanquart","doi":"10.1371/journal.pgen.1010842","DOIUrl":null,"url":null,"abstract":"<p><p>Escherichia coli is both a highly prevalent commensal and a major opportunistic pathogen causing bloodstream infections (BSI). A systematic analysis characterizing the genomic determinants of extra-intestinal pathogenic vs. commensal isolates in human populations, which could inform mechanisms of pathogenesis, diagnostic, prevention and treatment is still lacking. We used a collection of 912 BSI and 370 commensal E. coli isolates collected in France over a 17-year period (2000-2017). We compared their pangenomes, genetic backgrounds (phylogroups, STs, O groups), presence of virulence-associated genes (VAGs) and antimicrobial resistance genes, finding significant differences in all comparisons between commensal and BSI isolates. A machine learning linear model trained on all the genetic variants derived from the pangenome and controlling for population structure reveals similar differences in VAGs, discovers new variants associated with pathogenicity (capacity to cause BSI), and accurately classifies BSI vs. commensal strains. Pathogenicity is a highly heritable trait, with up to 69% of the variance explained by bacterial genetic variants. Lastly, complementing our commensal collection with an older collection from 1980, we predict that pathogenicity continuously increased through 1980, 2000, to 2010. Together our findings imply that E. coli exhibit substantial genetic variation contributing to the transition between commensalism and pathogenicity and that this species evolved towards higher pathogenicity.</p>","PeriodicalId":20266,"journal":{"name":"PLoS Genetics","volume":"19 8","pages":"e1010842"},"PeriodicalIF":3.7000,"publicationDate":"2023-08-02","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10395866/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"PLoS Genetics","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1371/journal.pgen.1010842","RegionNum":2,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2023/8/1 0:00:00","PubModel":"eCollection","JCR":"Q1","JCRName":"Agricultural and Biological Sciences","Score":null,"Total":0}
引用次数: 0
Abstract
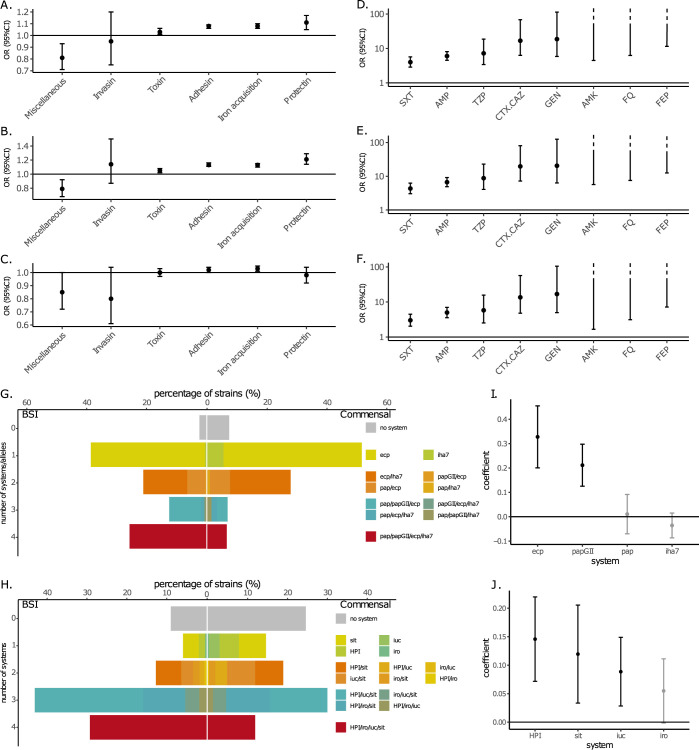
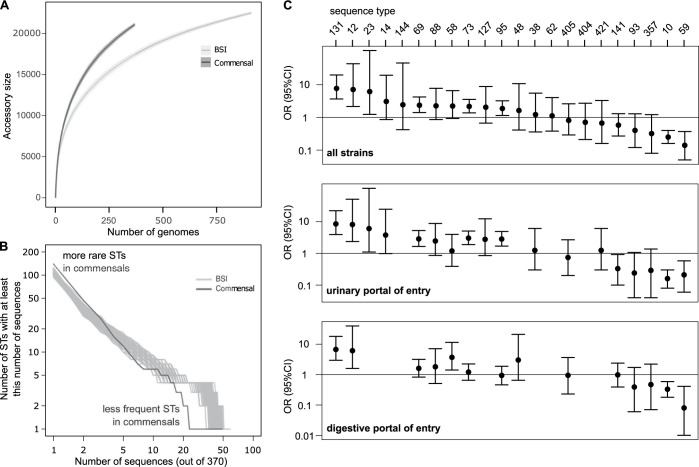
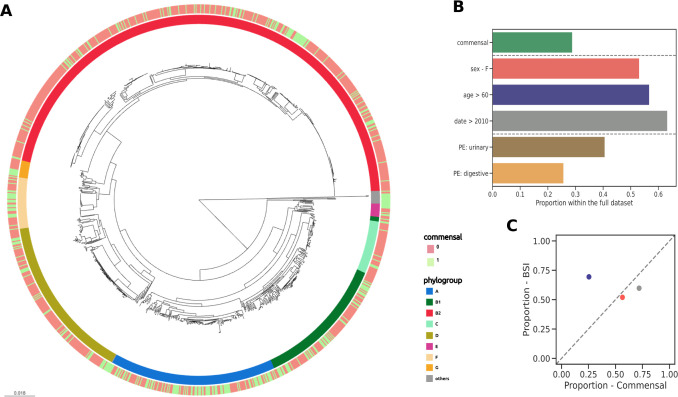
Escherichia coli is both a highly prevalent commensal and a major opportunistic pathogen causing bloodstream infections (BSI). A systematic analysis characterizing the genomic determinants of extra-intestinal pathogenic vs. commensal isolates in human populations, which could inform mechanisms of pathogenesis, diagnostic, prevention and treatment is still lacking. We used a collection of 912 BSI and 370 commensal E. coli isolates collected in France over a 17-year period (2000-2017). We compared their pangenomes, genetic backgrounds (phylogroups, STs, O groups), presence of virulence-associated genes (VAGs) and antimicrobial resistance genes, finding significant differences in all comparisons between commensal and BSI isolates. A machine learning linear model trained on all the genetic variants derived from the pangenome and controlling for population structure reveals similar differences in VAGs, discovers new variants associated with pathogenicity (capacity to cause BSI), and accurately classifies BSI vs. commensal strains. Pathogenicity is a highly heritable trait, with up to 69% of the variance explained by bacterial genetic variants. Lastly, complementing our commensal collection with an older collection from 1980, we predict that pathogenicity continuously increased through 1980, 2000, to 2010. Together our findings imply that E. coli exhibit substantial genetic variation contributing to the transition between commensalism and pathogenicity and that this species evolved towards higher pathogenicity.
期刊介绍:
PLOS Genetics is run by an international Editorial Board, headed by the Editors-in-Chief, Greg Barsh (HudsonAlpha Institute of Biotechnology, and Stanford University School of Medicine) and Greg Copenhaver (The University of North Carolina at Chapel Hill).
Articles published in PLOS Genetics are archived in PubMed Central and cited in PubMed.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
