L Sila, V Velmishi, B Saraci, E Dervishi, S Sila, D Shtiza, P Cullufi
{"title":"Congenital Hepatic Fibrosis as an Early Sign of Presentation of ADPKD.","authors":"L Sila, V Velmishi, B Saraci, E Dervishi, S Sila, D Shtiza, P Cullufi","doi":"10.2478/bjmg-2022-0024","DOIUrl":null,"url":null,"abstract":"<p><p>Autosomal dominant polycystic kidney disease (ADKPD) is the most frequent type of polycystic kidney disease. It is inherited through family members, with an incidence of approximately 1:400 to1:1000.Typically, individuals with ADKPD are identified between their fourth and fifth decade of life. ADKPD occurs as a results of mutation in one of the two genes, <i>PDK1</i> and <i>PDK2</i>.Patients with PKD1 experience renal failure at an earlier onset than those with PKD2. We report on a 2 year-old-boy with hepatosplenomegaly and signs of portal hypertension. Both kidneys appeared normal until the age of 8, when multiple cysts developed, this being typical of ADKPD. Suspecting ADKPD, we performed whole exome sequencing, thereby confirming a mutation of c.6730 673del p.(Ser 2244Hisfs*17). The investigations of all family members found other individuals affected by ADKPD.</p>","PeriodicalId":55403,"journal":{"name":"Balkan Journal of Medical Genetics","volume":"25 2","pages":"91-95"},"PeriodicalIF":0.9000,"publicationDate":"2023-05-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/f3/ba/bjmg-25-2-bjmg-2022-0024.PMC10230838.pdf","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Balkan Journal of Medical Genetics","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.2478/bjmg-2022-0024","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q4","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
引用次数: 0
Abstract
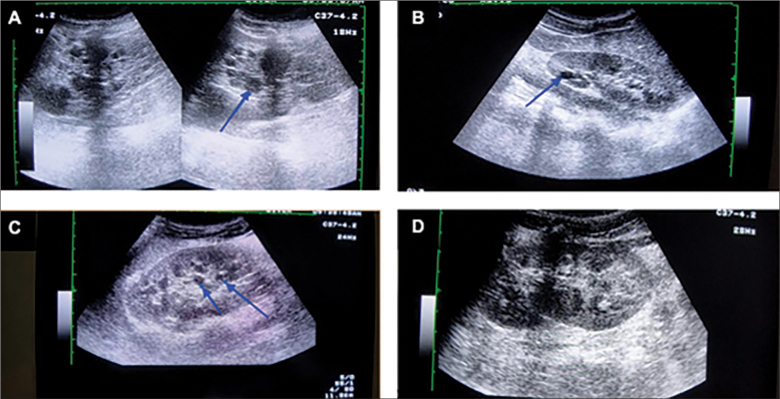
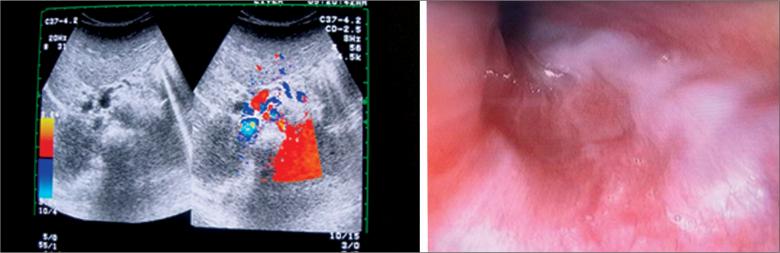
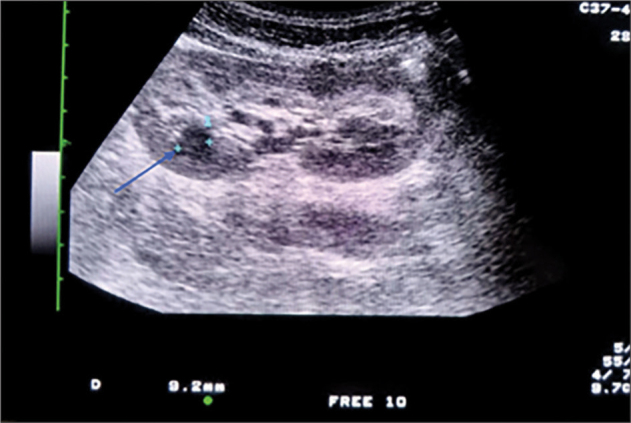
Autosomal dominant polycystic kidney disease (ADKPD) is the most frequent type of polycystic kidney disease. It is inherited through family members, with an incidence of approximately 1:400 to1:1000.Typically, individuals with ADKPD are identified between their fourth and fifth decade of life. ADKPD occurs as a results of mutation in one of the two genes, PDK1 and PDK2.Patients with PKD1 experience renal failure at an earlier onset than those with PKD2. We report on a 2 year-old-boy with hepatosplenomegaly and signs of portal hypertension. Both kidneys appeared normal until the age of 8, when multiple cysts developed, this being typical of ADKPD. Suspecting ADKPD, we performed whole exome sequencing, thereby confirming a mutation of c.6730 673del p.(Ser 2244Hisfs*17). The investigations of all family members found other individuals affected by ADKPD.
期刊介绍:
Balkan Journal of Medical Genetics is a journal in the English language for publication of articles involving all branches of medical genetics: human cytogenetics, molecular genetics, clinical genetics, immunogenetics, oncogenetics, pharmacogenetics, population genetics, genetic screening and diagnosis of monogenic and polygenic diseases, prenatal and preimplantation genetic diagnosis, genetic counselling, advances in treatment and prevention.


 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
