N Sreedevi, N Swapna, Santosh Maruthy, T Jayakumar, Charles Sylvester
{"title":"Molecular Evaluation of Joubert Syndrome and Hearing Impairment in a Patient with Ataxic Cerebral Palsy.","authors":"N Sreedevi, N Swapna, Santosh Maruthy, T Jayakumar, Charles Sylvester","doi":"10.1055/s-0043-1771184","DOIUrl":null,"url":null,"abstract":"<p><p>Joubert syndrome (JBTS) is a rare autosomal recessive or X-linked congenital brain malformation with strong genetic heterogeneity. Other neurological features of JBTS include hypotonia, ataxia, developmental delay, and cognitive impairment. Hearing loss with JBTS has been reported in the literature. We present the case of a 3.5-year-old boy born to a healthy consanguineous South Indian couple who was presented with ataxic cerebral palsy (CP) and hearing impairment; medical reports confirmed typical brain malformations of JBTS. Hearing impairment was screened by audiological assessment, which confirmed the presence of severe-profound hearing loss with outer hair cell dysfunction. Whole-exome sequencing (WES) was performed to know the molecular aspects of the condition and to detect any novel mutations. The homozygous mutation <i>AHI1</i> c.2023G > A associated with JBTS type 3 and <i>GJB2</i> c.71G > A mutation associated with hearing impairment were identified. Sanger sequencing was performed to validate the result and it identified heterozygous <i>AHI1</i> c.2023G > A and <i>GJB2</i> c.71G > A in the patient's parents. This study confirms the diagnosis of JBTS by WES helps identify the genetic causes of hereditary disorders that accelerate genetic evaluation and counseling for at-risk families.</p>","PeriodicalId":40142,"journal":{"name":"Global Medical Genetics","volume":"10 3","pages":"190-193"},"PeriodicalIF":1.5000,"publicationDate":"2023-09-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10370468/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Global Medical Genetics","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1055/s-0043-1771184","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q4","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
引用次数: 0
Abstract
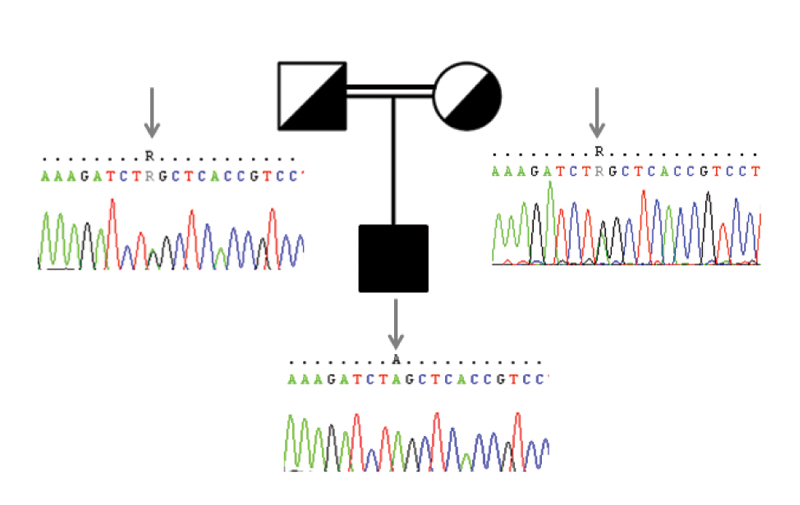
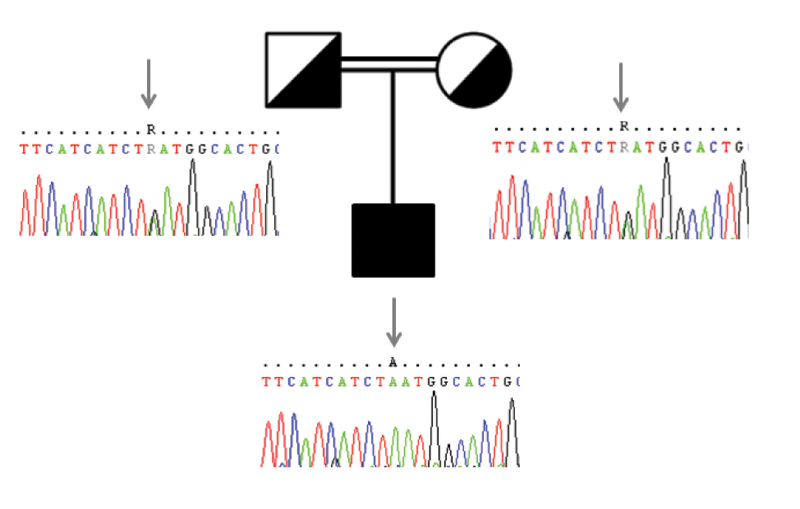
Joubert syndrome (JBTS) is a rare autosomal recessive or X-linked congenital brain malformation with strong genetic heterogeneity. Other neurological features of JBTS include hypotonia, ataxia, developmental delay, and cognitive impairment. Hearing loss with JBTS has been reported in the literature. We present the case of a 3.5-year-old boy born to a healthy consanguineous South Indian couple who was presented with ataxic cerebral palsy (CP) and hearing impairment; medical reports confirmed typical brain malformations of JBTS. Hearing impairment was screened by audiological assessment, which confirmed the presence of severe-profound hearing loss with outer hair cell dysfunction. Whole-exome sequencing (WES) was performed to know the molecular aspects of the condition and to detect any novel mutations. The homozygous mutation AHI1 c.2023G > A associated with JBTS type 3 and GJB2 c.71G > A mutation associated with hearing impairment were identified. Sanger sequencing was performed to validate the result and it identified heterozygous AHI1 c.2023G > A and GJB2 c.71G > A in the patient's parents. This study confirms the diagnosis of JBTS by WES helps identify the genetic causes of hereditary disorders that accelerate genetic evaluation and counseling for at-risk families.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
