Bo Wang, Qian Zhang, Lili Wu, Cunliang Deng, Meiyan Luo, Yu Xie, Gang Wu, Wen Chen, Yunjian Sheng, Peng Zhu, Gang Qin
{"title":"Data-independent acquisition-based mass spectrometry(DIA-MS) for quantitative analysis of patients with chronic hepatitis B.","authors":"Bo Wang, Qian Zhang, Lili Wu, Cunliang Deng, Meiyan Luo, Yu Xie, Gang Wu, Wen Chen, Yunjian Sheng, Peng Zhu, Gang Qin","doi":"10.1186/s12953-023-00209-6","DOIUrl":null,"url":null,"abstract":"<p><p>Chronic hepatitis B is a significant public health problem and complex pathologic process, and unraveling the underlying mechanisms and pathophysiology is of great significance. Data independent acquisition mass spectrometry (DIA-MS) is a label-free quantitative proteomics method that has been successfully applied to the study of a wide range of diseases. The aim of this study was to apply DIA-MS for proteomic analysis of patients with chronic hepatitis B. We performed comprehensive proteomics analysis of protein expression in serum samples from HBV patients and healthy controls by using DIA-MS. Gene Ontology (GO) terms, Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways, and protein network analysis were performed on differentially expressed proteins and were further combined with literature analysis. We successfully identified a total of 3786 serum proteins with a high quantitative performance from serum samples in this study. We identified 310 differentially expressed proteins (DEPs) (fold change > 1.5 and P value < 0.05 as the criteria for a significant difference) between HBV and healthy samples. A total of 242 upregulated proteins and 68 downregulated proteins were among the DEPs. Some protein expression levels were significantly elevated or decreased in patients with chronic hepatitis B, indicating a relation to chronic liver disease, which should be further investigated.</p>","PeriodicalId":20857,"journal":{"name":"Proteome Science","volume":"21 1","pages":"9"},"PeriodicalIF":1.6000,"publicationDate":"2023-06-06","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10246044/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Proteome Science","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1186/s12953-023-00209-6","RegionNum":3,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"BIOCHEMICAL RESEARCH METHODS","Score":null,"Total":0}
引用次数: 0
Abstract
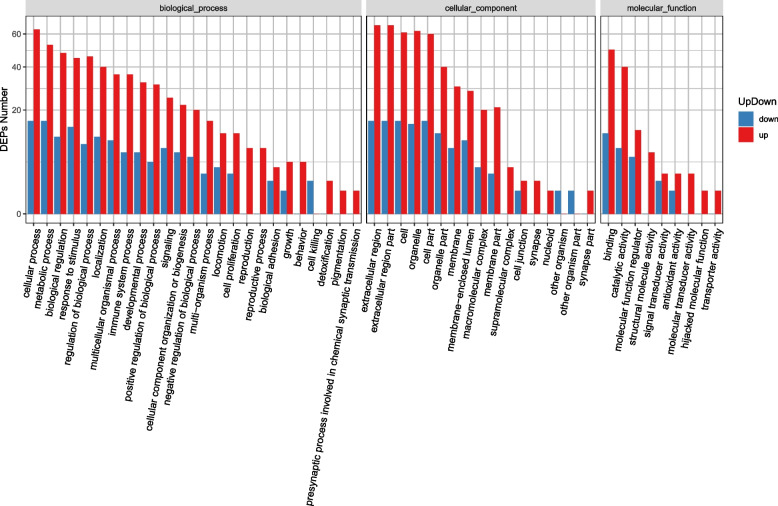
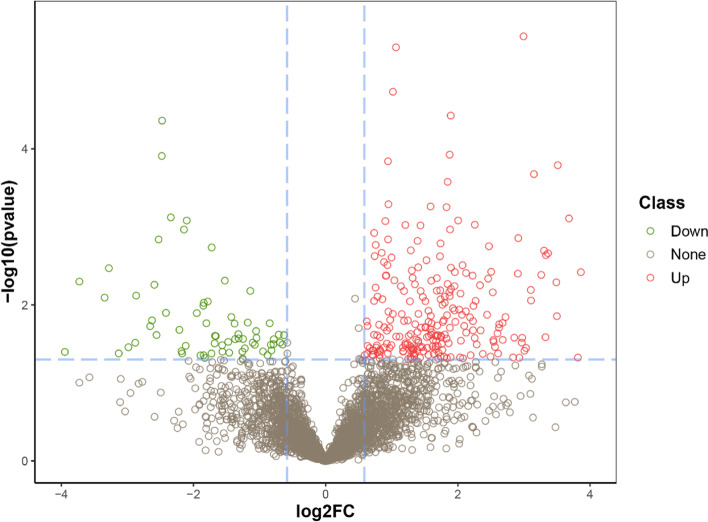
Chronic hepatitis B is a significant public health problem and complex pathologic process, and unraveling the underlying mechanisms and pathophysiology is of great significance. Data independent acquisition mass spectrometry (DIA-MS) is a label-free quantitative proteomics method that has been successfully applied to the study of a wide range of diseases. The aim of this study was to apply DIA-MS for proteomic analysis of patients with chronic hepatitis B. We performed comprehensive proteomics analysis of protein expression in serum samples from HBV patients and healthy controls by using DIA-MS. Gene Ontology (GO) terms, Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways, and protein network analysis were performed on differentially expressed proteins and were further combined with literature analysis. We successfully identified a total of 3786 serum proteins with a high quantitative performance from serum samples in this study. We identified 310 differentially expressed proteins (DEPs) (fold change > 1.5 and P value < 0.05 as the criteria for a significant difference) between HBV and healthy samples. A total of 242 upregulated proteins and 68 downregulated proteins were among the DEPs. Some protein expression levels were significantly elevated or decreased in patients with chronic hepatitis B, indicating a relation to chronic liver disease, which should be further investigated.
期刊介绍:
Proteome Science is an open access journal publishing research in the area of systems studies. Proteome Science considers manuscripts based on all aspects of functional and structural proteomics, genomics, metabolomics, systems analysis and metabiome analysis. It encourages the submissions of studies that use large-scale or systems analysis of biomolecules in a cellular, organismal and/or environmental context.
Studies that describe novel biological or clinical insights as well as methods-focused studies that describe novel methods for the large-scale study of any and all biomolecules in cells and tissues, such as mass spectrometry, protein and nucleic acid microarrays, genomics, next-generation sequencing and computational algorithms and methods are all within the scope of Proteome Science, as are electron topography, structural methods, proteogenomics, chemical proteomics, stem cell proteomics, organelle proteomics, plant and microbial proteomics.
In spite of its name, Proteome Science considers all aspects of large-scale and systems studies because ultimately any mechanism that results in genomic and metabolomic changes will affect or be affected by the proteome. To reflect this intrinsic relationship of biological systems, Proteome Science will consider all such articles.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
