Myelin oligodendrocyte glycoprotein antibody-associated disease as a novel presentation of central nervous system autoimmunity in a pediatric patient with Wiskott-Aldrich syndrome.
Vivien X Xie, Wilson File, Christina Wiedl, Brant R Ward, Blachy Dávila Saldaña, Michael D Keller, Alexandra B Kornbluh
{"title":"Myelin oligodendrocyte glycoprotein antibody-associated disease as a novel presentation of central nervous system autoimmunity in a pediatric patient with Wiskott-Aldrich syndrome.","authors":"Vivien X Xie, Wilson File, Christina Wiedl, Brant R Ward, Blachy Dávila Saldaña, Michael D Keller, Alexandra B Kornbluh","doi":"10.1186/s13223-023-00827-x","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Wiskott-Aldrich syndrome (WAS) is an X-linked primary immunodeficiency caused by mutations in the WAS gene that leads to increased susceptibility to infections, thrombocytopenia, eczema, malignancies, and autoimmunity. Central nervous system (CNS) autoimmune manifestations are uncommon.</p><p><strong>Case presentation: </strong>We describe the case of a five-year-old boy with refractory thrombocytopenia and iron deficiency anemia who developed relapsing bilateral optic neuritis. Myelin oligodendrocyte glycoprotein antibody (MOG-IgG) via serum fluorescence-activated cell sorting assay was positive (titer 1:100), confirming a diagnosis of myelin oligodendrocyte glycoprotein antibody-associated disease (MOGAD). At age six, molecular panel testing for genes associated with primary immunodeficiency identified a missense WAS gene variant. He was subsequently found to have decreased WAS protein expression, consistent with a diagnosis of WAS.</p><p><strong>Conclusions: </strong>This case expands the reported spectrum of CNS autoimmunity associated with WAS and may help to inform long-term therapeutic options.</p>","PeriodicalId":7702,"journal":{"name":"Allergy, Asthma, and Clinical Immunology : Official Journal of the Canadian Society of Allergy and Clinical Immunology","volume":"19 1","pages":"68"},"PeriodicalIF":0.0000,"publicationDate":"2023-08-07","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10408201/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Allergy, Asthma, and Clinical Immunology : Official Journal of the Canadian Society of Allergy and Clinical Immunology","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1186/s13223-023-00827-x","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 0
Abstract
Background: Wiskott-Aldrich syndrome (WAS) is an X-linked primary immunodeficiency caused by mutations in the WAS gene that leads to increased susceptibility to infections, thrombocytopenia, eczema, malignancies, and autoimmunity. Central nervous system (CNS) autoimmune manifestations are uncommon.
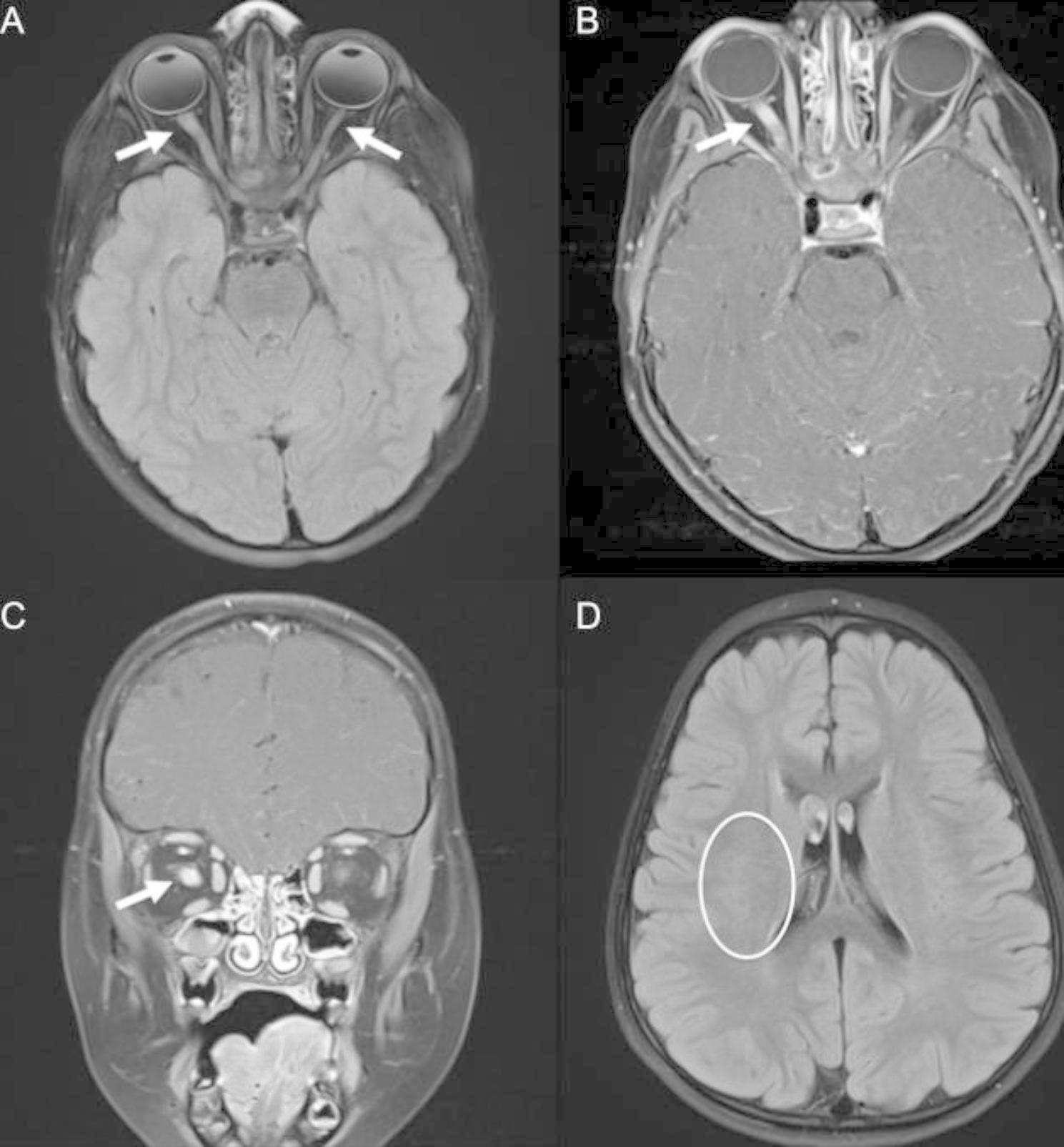
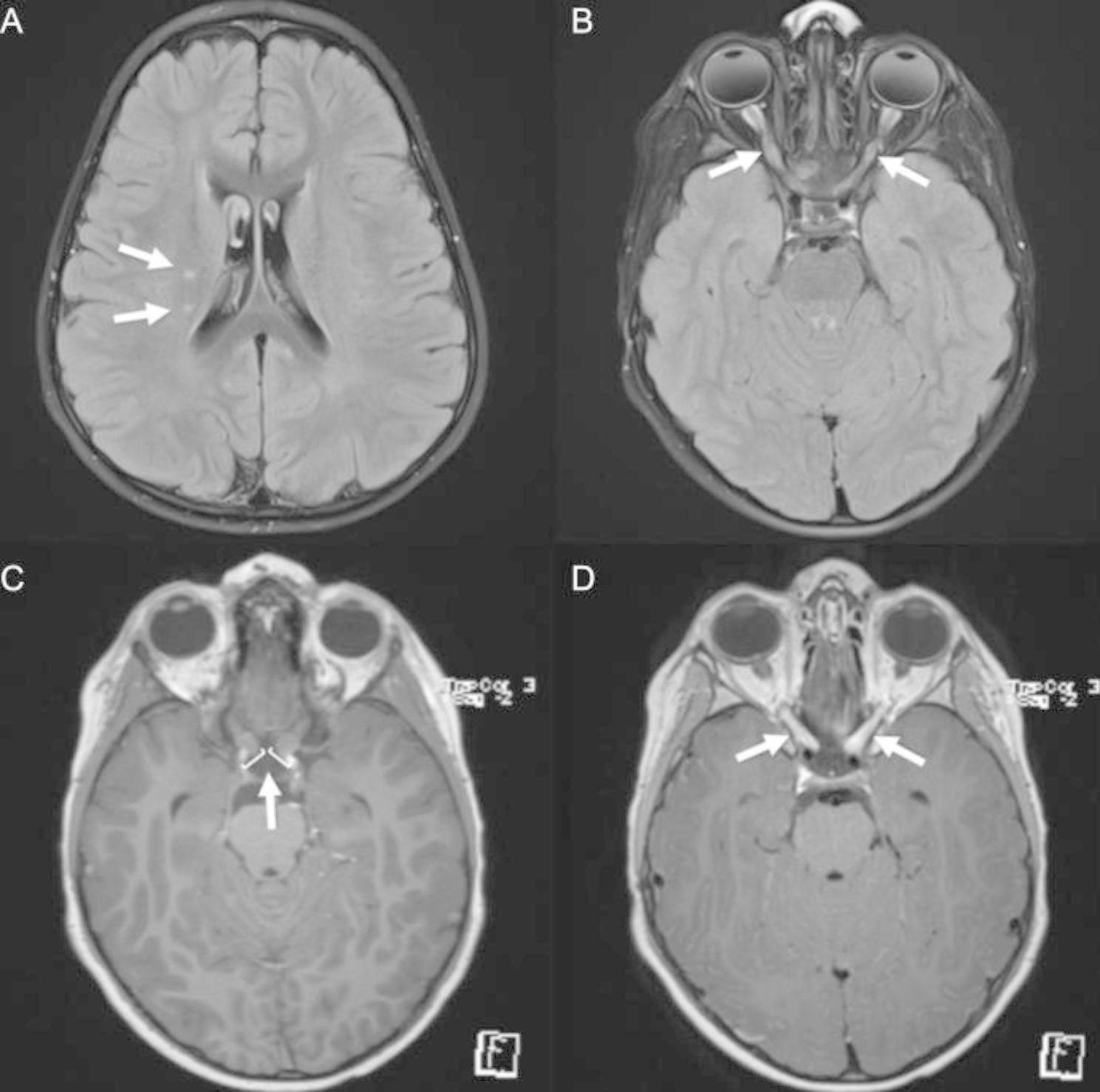
Case presentation: We describe the case of a five-year-old boy with refractory thrombocytopenia and iron deficiency anemia who developed relapsing bilateral optic neuritis. Myelin oligodendrocyte glycoprotein antibody (MOG-IgG) via serum fluorescence-activated cell sorting assay was positive (titer 1:100), confirming a diagnosis of myelin oligodendrocyte glycoprotein antibody-associated disease (MOGAD). At age six, molecular panel testing for genes associated with primary immunodeficiency identified a missense WAS gene variant. He was subsequently found to have decreased WAS protein expression, consistent with a diagnosis of WAS.
Conclusions: This case expands the reported spectrum of CNS autoimmunity associated with WAS and may help to inform long-term therapeutic options.


 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
