Ryan S McClure, Yvonne Rericha, Katrina M Waters, Robyn L Tanguay
{"title":"3' RNA-seq is superior to standard RNA-seq in cases of sparse data but inferior at identifying toxicity pathways in a model organism.","authors":"Ryan S McClure, Yvonne Rericha, Katrina M Waters, Robyn L Tanguay","doi":"10.3389/fbinf.2023.1234218","DOIUrl":null,"url":null,"abstract":"<p><p><b>Introduction:</b> The application of RNA-sequencing has led to numerous breakthroughs related to investigating gene expression levels in complex biological systems. Among these are knowledge of how organisms, such as the vertebrate model organism zebrafish (<i>Danio rerio</i>), respond to toxicant exposure. Recently, the development of 3' RNA-seq has allowed for the determination of gene expression levels with a fraction of the required reads compared to standard RNA-seq. While 3' RNA-seq has many advantages, a comparison to standard RNA-seq has not been performed in the context of whole organism toxicity and sparse data. <b>Methods and results:</b> Here, we examined samples from zebrafish exposed to perfluorobutane sulfonamide (FBSA) with either 3' or standard RNA-seq to determine the advantages of each with regards to the identification of functionally enriched pathways. We found that 3' and standard RNA-seq showed specific advantages when focusing on annotated or unannotated regions of the genome. We also found that standard RNA-seq identified more differentially expressed genes (DEGs), but that this advantage disappeared under conditions of sparse data. We also found that standard RNA-seq had a significant advantage in identifying functionally enriched pathways via analysis of DEG lists but that this advantage was minimal when identifying pathways via gene set enrichment analysis of all genes. <b>Conclusions:</b> These results show that each approach has experimental conditions where they may be advantageous. Our observations can help guide others in the choice of 3' RNA-seq vs standard RNA sequencing to query gene expression levels in a range of biological systems.</p>","PeriodicalId":73066,"journal":{"name":"Frontiers in bioinformatics","volume":"3 ","pages":"1234218"},"PeriodicalIF":3.9000,"publicationDate":"2023-07-27","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10414111/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Frontiers in bioinformatics","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.3389/fbinf.2023.1234218","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2023/1/1 0:00:00","PubModel":"eCollection","JCR":"Q2","JCRName":"MATHEMATICAL & COMPUTATIONAL BIOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
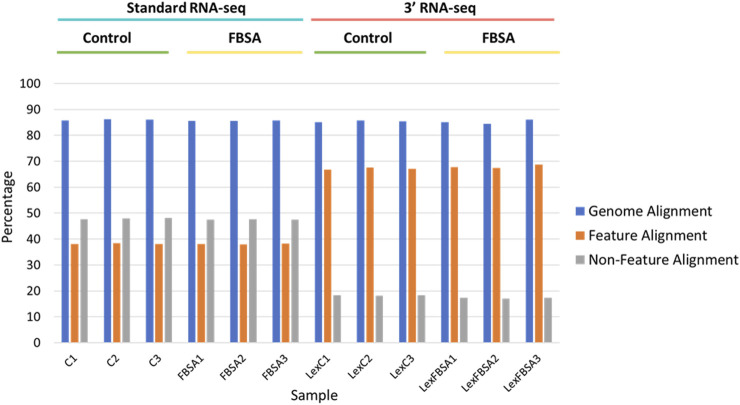
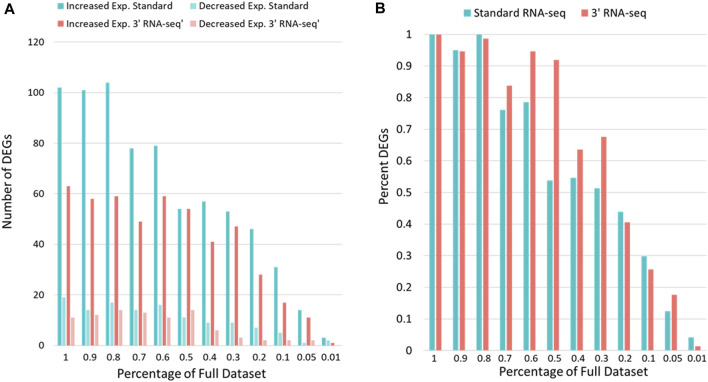
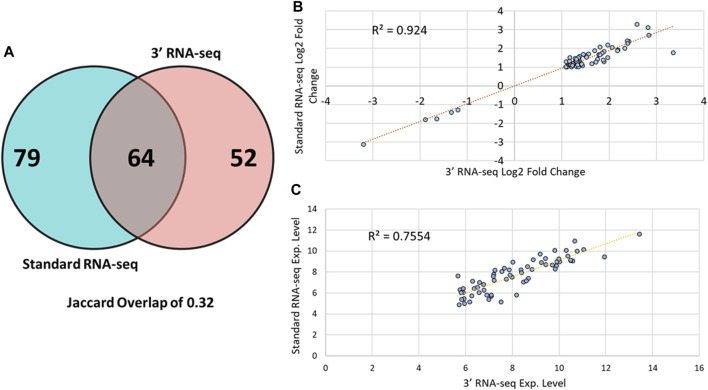
Introduction: The application of RNA-sequencing has led to numerous breakthroughs related to investigating gene expression levels in complex biological systems. Among these are knowledge of how organisms, such as the vertebrate model organism zebrafish (Danio rerio), respond to toxicant exposure. Recently, the development of 3' RNA-seq has allowed for the determination of gene expression levels with a fraction of the required reads compared to standard RNA-seq. While 3' RNA-seq has many advantages, a comparison to standard RNA-seq has not been performed in the context of whole organism toxicity and sparse data. Methods and results: Here, we examined samples from zebrafish exposed to perfluorobutane sulfonamide (FBSA) with either 3' or standard RNA-seq to determine the advantages of each with regards to the identification of functionally enriched pathways. We found that 3' and standard RNA-seq showed specific advantages when focusing on annotated or unannotated regions of the genome. We also found that standard RNA-seq identified more differentially expressed genes (DEGs), but that this advantage disappeared under conditions of sparse data. We also found that standard RNA-seq had a significant advantage in identifying functionally enriched pathways via analysis of DEG lists but that this advantage was minimal when identifying pathways via gene set enrichment analysis of all genes. Conclusions: These results show that each approach has experimental conditions where they may be advantageous. Our observations can help guide others in the choice of 3' RNA-seq vs standard RNA sequencing to query gene expression levels in a range of biological systems.


 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
