Jeffrey C Y Yu, Yixiao Zeng, Kaiqiong Zhao, Tianyuan Lu, Kathleen Oros Klein, Inés Colmegna, Maximilien Lora, Sahir R Bhatnagar, Andrew Leask, Celia M T Greenwood, Marie Hudson
{"title":"利用灵敏的计算方法分析全基因组亚硫酸氢盐测序数据,对系统性硬化症有了新的认识。","authors":"Jeffrey C Y Yu, Yixiao Zeng, Kaiqiong Zhao, Tianyuan Lu, Kathleen Oros Klein, Inés Colmegna, Maximilien Lora, Sahir R Bhatnagar, Andrew Leask, Celia M T Greenwood, Marie Hudson","doi":"10.1186/s13148-023-01513-w","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Abnormal DNA methylation is thought to contribute to the onset and progression of systemic sclerosis. Currently, the most comprehensive assay for profiling DNA methylation is whole-genome bisulfite sequencing (WGBS), but its precision depends on read depth and it may be subject to sequencing errors. SOMNiBUS, a method for regional analysis, attempts to overcome some of these limitations. Using SOMNiBUS, we re-analyzed WGBS data previously analyzed using bumphunter, an approach that initially fits single CpG associations, to contrast DNA methylation estimates by both methods.</p><p><strong>Methods: </strong>Purified CD4+ T lymphocytes of 9 SSc and 4 control females were sequenced using WGBS. We separated the resulting sequencing data into regions with dense CpG data, and differentially methylated regions (DMRs) were inferred with the SOMNiBUS region-level test, adjusted for age. Pathway enrichment analysis was performed with ingenuity pathway analysis (IPA). We compared the results obtained by SOMNiBUS and bumphunter.</p><p><strong>Results: </strong>Of 8268 CpG regions of ≥ 60 CpGs eligible for analysis with SOMNiBUS, we identified 131 DMRs and 125 differentially methylated genes (DMGs; p-values less than Bonferroni-corrected threshold of 6.05-06 controlling family-wise error rate at 0.05; 1.6% of the regions). In comparison, bumphunter identified 821,929 CpG regions, 599 DMRs (of which none had ≥ 60 CpGs) and 340 DMGs (q-value of 0.05; 0.04% of all regions). The top ranked gene identified by SOMNiBUS was FLT4, a lymphangiogenic orchestrator, and the top ranked gene on chromosome X was CHST7, known to catalyze the sulfation of glycosaminoglycans in the extracellular matrix. The top networks identified by IPA included connective tissue disorders.</p><p><strong>Conclusions: </strong>SOMNiBUS is a complementary method of analyzing WGBS data that enhances biological insights into SSc and provides novel avenues of investigation into its pathogenesis.</p>","PeriodicalId":48652,"journal":{"name":"Clinical Epigenetics","volume":"15 1","pages":"96"},"PeriodicalIF":4.4000,"publicationDate":"2023-06-03","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10239181/pdf/","citationCount":"0","resultStr":"{\"title\":\"Novel insights into systemic sclerosis using a sensitive computational method to analyze whole-genome bisulfite sequencing data.\",\"authors\":\"Jeffrey C Y Yu, Yixiao Zeng, Kaiqiong Zhao, Tianyuan Lu, Kathleen Oros Klein, Inés Colmegna, Maximilien Lora, Sahir R Bhatnagar, Andrew Leask, Celia M T Greenwood, Marie Hudson\",\"doi\":\"10.1186/s13148-023-01513-w\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Background: </strong>Abnormal DNA methylation is thought to contribute to the onset and progression of systemic sclerosis. Currently, the most comprehensive assay for profiling DNA methylation is whole-genome bisulfite sequencing (WGBS), but its precision depends on read depth and it may be subject to sequencing errors. SOMNiBUS, a method for regional analysis, attempts to overcome some of these limitations. Using SOMNiBUS, we re-analyzed WGBS data previously analyzed using bumphunter, an approach that initially fits single CpG associations, to contrast DNA methylation estimates by both methods.</p><p><strong>Methods: </strong>Purified CD4+ T lymphocytes of 9 SSc and 4 control females were sequenced using WGBS. We separated the resulting sequencing data into regions with dense CpG data, and differentially methylated regions (DMRs) were inferred with the SOMNiBUS region-level test, adjusted for age. Pathway enrichment analysis was performed with ingenuity pathway analysis (IPA). We compared the results obtained by SOMNiBUS and bumphunter.</p><p><strong>Results: </strong>Of 8268 CpG regions of ≥ 60 CpGs eligible for analysis with SOMNiBUS, we identified 131 DMRs and 125 differentially methylated genes (DMGs; p-values less than Bonferroni-corrected threshold of 6.05-06 controlling family-wise error rate at 0.05; 1.6% of the regions). In comparison, bumphunter identified 821,929 CpG regions, 599 DMRs (of which none had ≥ 60 CpGs) and 340 DMGs (q-value of 0.05; 0.04% of all regions). The top ranked gene identified by SOMNiBUS was FLT4, a lymphangiogenic orchestrator, and the top ranked gene on chromosome X was CHST7, known to catalyze the sulfation of glycosaminoglycans in the extracellular matrix. The top networks identified by IPA included connective tissue disorders.</p><p><strong>Conclusions: </strong>SOMNiBUS is a complementary method of analyzing WGBS data that enhances biological insights into SSc and provides novel avenues of investigation into its pathogenesis.</p>\",\"PeriodicalId\":48652,\"journal\":{\"name\":\"Clinical Epigenetics\",\"volume\":\"15 1\",\"pages\":\"96\"},\"PeriodicalIF\":4.4000,\"publicationDate\":\"2023-06-03\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10239181/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Clinical Epigenetics\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://doi.org/10.1186/s13148-023-01513-w\",\"RegionNum\":2,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"Medicine\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Clinical Epigenetics","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1186/s13148-023-01513-w","RegionNum":2,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"Medicine","Score":null,"Total":0}
Novel insights into systemic sclerosis using a sensitive computational method to analyze whole-genome bisulfite sequencing data.
Background: Abnormal DNA methylation is thought to contribute to the onset and progression of systemic sclerosis. Currently, the most comprehensive assay for profiling DNA methylation is whole-genome bisulfite sequencing (WGBS), but its precision depends on read depth and it may be subject to sequencing errors. SOMNiBUS, a method for regional analysis, attempts to overcome some of these limitations. Using SOMNiBUS, we re-analyzed WGBS data previously analyzed using bumphunter, an approach that initially fits single CpG associations, to contrast DNA methylation estimates by both methods.
Methods: Purified CD4+ T lymphocytes of 9 SSc and 4 control females were sequenced using WGBS. We separated the resulting sequencing data into regions with dense CpG data, and differentially methylated regions (DMRs) were inferred with the SOMNiBUS region-level test, adjusted for age. Pathway enrichment analysis was performed with ingenuity pathway analysis (IPA). We compared the results obtained by SOMNiBUS and bumphunter.
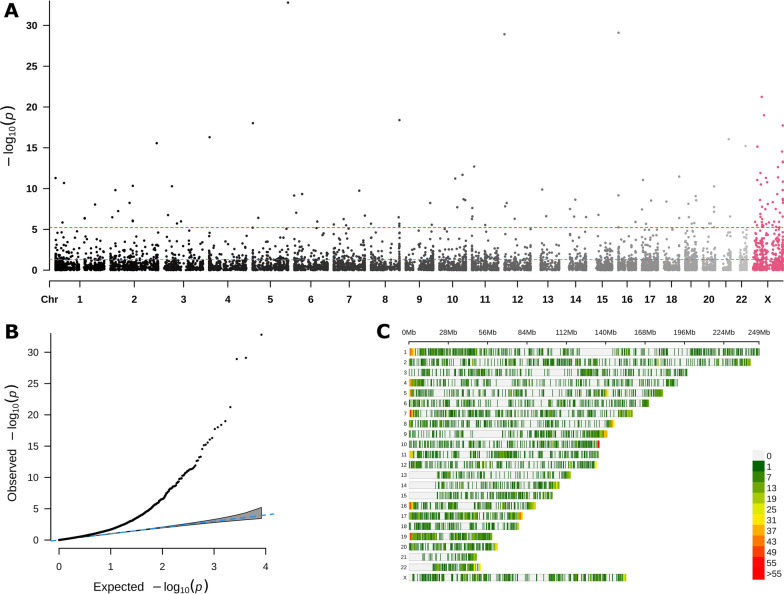
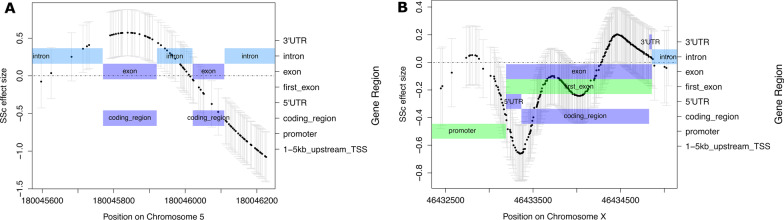
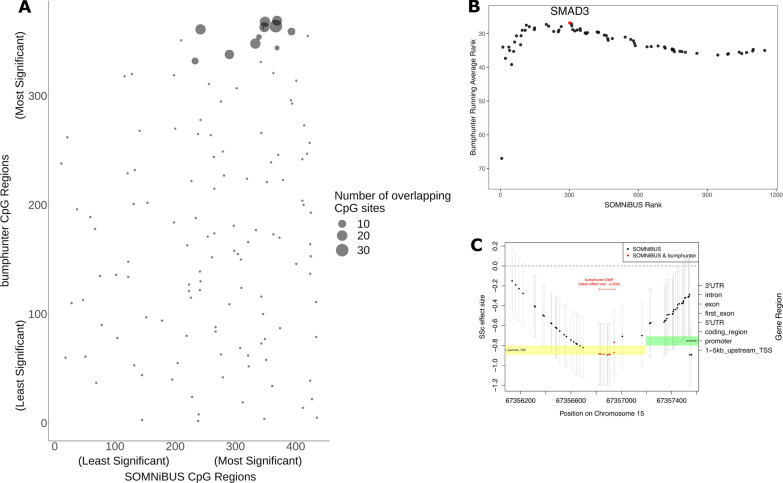
Results: Of 8268 CpG regions of ≥ 60 CpGs eligible for analysis with SOMNiBUS, we identified 131 DMRs and 125 differentially methylated genes (DMGs; p-values less than Bonferroni-corrected threshold of 6.05-06 controlling family-wise error rate at 0.05; 1.6% of the regions). In comparison, bumphunter identified 821,929 CpG regions, 599 DMRs (of which none had ≥ 60 CpGs) and 340 DMGs (q-value of 0.05; 0.04% of all regions). The top ranked gene identified by SOMNiBUS was FLT4, a lymphangiogenic orchestrator, and the top ranked gene on chromosome X was CHST7, known to catalyze the sulfation of glycosaminoglycans in the extracellular matrix. The top networks identified by IPA included connective tissue disorders.
Conclusions: SOMNiBUS is a complementary method of analyzing WGBS data that enhances biological insights into SSc and provides novel avenues of investigation into its pathogenesis.
Clinical EpigeneticsBiochemistry, Genetics and Molecular Biology-Developmental Biology
CiteScore
8.90
自引率
5.30%
发文量
150
审稿时长
12 weeks
期刊介绍:
Clinical Epigenetics, the official journal of the Clinical Epigenetics Society, is an open access, peer-reviewed journal that encompasses all aspects of epigenetic principles and mechanisms in relation to human disease, diagnosis and therapy. Clinical trials and research in disease model organisms are particularly welcome.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
