{"title":"高磷血症家族性肿瘤性钙质沉着症,由于母亲单亲二体GALNT3变异体。","authors":"Naoko Nishimura-Kinoshita, Yasuhisa Ohata, Hiromi Sawai, Masako Izawa, Shinji Takeyari, Takuo Kubota, Yosuke Omae, Keiichi Ozono, Katsushi Tokunaga, Takashi Hamajima","doi":"10.1297/cpe.2022-0071","DOIUrl":null,"url":null,"abstract":"<p><p>Hyperphosphatemic familial tumoral calcinosis (HFTC) is a rare, inherited autosomal recessive disorder caused by fibroblast growth factor-23 (<i>FGF23</i>), N-acetylgalactosaminyltransferase 3 (<i>GALNT3</i>), or Klotho (<i>KL</i>) gene variants. Here, we report the case of a Japanese boy who presented with a mass in his left elbow at the age of three. Laboratory test results of the patient revealed normocalcemia (10.3 mg/dL) and hyperphosphatemia (8.7 mg/dL); however, despite hyperphosphatemia, serum intact FGF23 level was low, renal tubular reabsorption of phosphate (TRP) level was inappropriately increased, and 1,25-dihydroxyvitamin D<sub>3</sub> (1,25(OH)<sub>2</sub>D<sub>3</sub>) level was inappropriately normal. Genetic analysis revealed maternal uniparental disomy (UPD) of chromosome 2, which included a novel <i>GALNT3</i> variant (c.1780-1G>C). Reverse transcription-polymerase chain reaction (RT-PCR) analysis of <i>GALNT3</i> mRNA confirmed that this variant resulted in the destruction of exon 11. We resected the mass when the patient was five years old, owing to its gradual enlargement. No relapse or new pathological lesions were observed four years after tumor resection. This is the first case report of a Japanese patient with HFTC associated with a novel <i>GALNT3</i> variant, as well as the first case of HFTC caused by maternal UPD of chromosome 2 that includes the <i>GALNT3</i> variant.</p>","PeriodicalId":72619,"journal":{"name":"","volume":"32 3","pages":"161-167"},"PeriodicalIF":0.0,"publicationDate":"2023-01-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/34/62/cpe-32-161.PMC10288290.pdf","citationCount":"0","resultStr":"{\"title\":\"A case of hyperphosphatemic familial tumoral calcinosis due to maternal uniparental disomy of a <i>GALNT3</i> variant.\",\"authors\":\"Naoko Nishimura-Kinoshita, Yasuhisa Ohata, Hiromi Sawai, Masako Izawa, Shinji Takeyari, Takuo Kubota, Yosuke Omae, Keiichi Ozono, Katsushi Tokunaga, Takashi Hamajima\",\"doi\":\"10.1297/cpe.2022-0071\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Hyperphosphatemic familial tumoral calcinosis (HFTC) is a rare, inherited autosomal recessive disorder caused by fibroblast growth factor-23 (<i>FGF23</i>), N-acetylgalactosaminyltransferase 3 (<i>GALNT3</i>), or Klotho (<i>KL</i>) gene variants. Here, we report the case of a Japanese boy who presented with a mass in his left elbow at the age of three. Laboratory test results of the patient revealed normocalcemia (10.3 mg/dL) and hyperphosphatemia (8.7 mg/dL); however, despite hyperphosphatemia, serum intact FGF23 level was low, renal tubular reabsorption of phosphate (TRP) level was inappropriately increased, and 1,25-dihydroxyvitamin D<sub>3</sub> (1,25(OH)<sub>2</sub>D<sub>3</sub>) level was inappropriately normal. Genetic analysis revealed maternal uniparental disomy (UPD) of chromosome 2, which included a novel <i>GALNT3</i> variant (c.1780-1G>C). Reverse transcription-polymerase chain reaction (RT-PCR) analysis of <i>GALNT3</i> mRNA confirmed that this variant resulted in the destruction of exon 11. We resected the mass when the patient was five years old, owing to its gradual enlargement. No relapse or new pathological lesions were observed four years after tumor resection. This is the first case report of a Japanese patient with HFTC associated with a novel <i>GALNT3</i> variant, as well as the first case of HFTC caused by maternal UPD of chromosome 2 that includes the <i>GALNT3</i> variant.</p>\",\"PeriodicalId\":72619,\"journal\":{\"name\":\"\",\"volume\":\"32 3\",\"pages\":\"161-167\"},\"PeriodicalIF\":0.0,\"publicationDate\":\"2023-01-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/34/62/cpe-32-161.PMC10288290.pdf\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1297/cpe.2022-0071\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"\",\"JCRName\":\"\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1297/cpe.2022-0071","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"","JCRName":"","Score":null,"Total":0}
A case of hyperphosphatemic familial tumoral calcinosis due to maternal uniparental disomy of a GALNT3 variant.
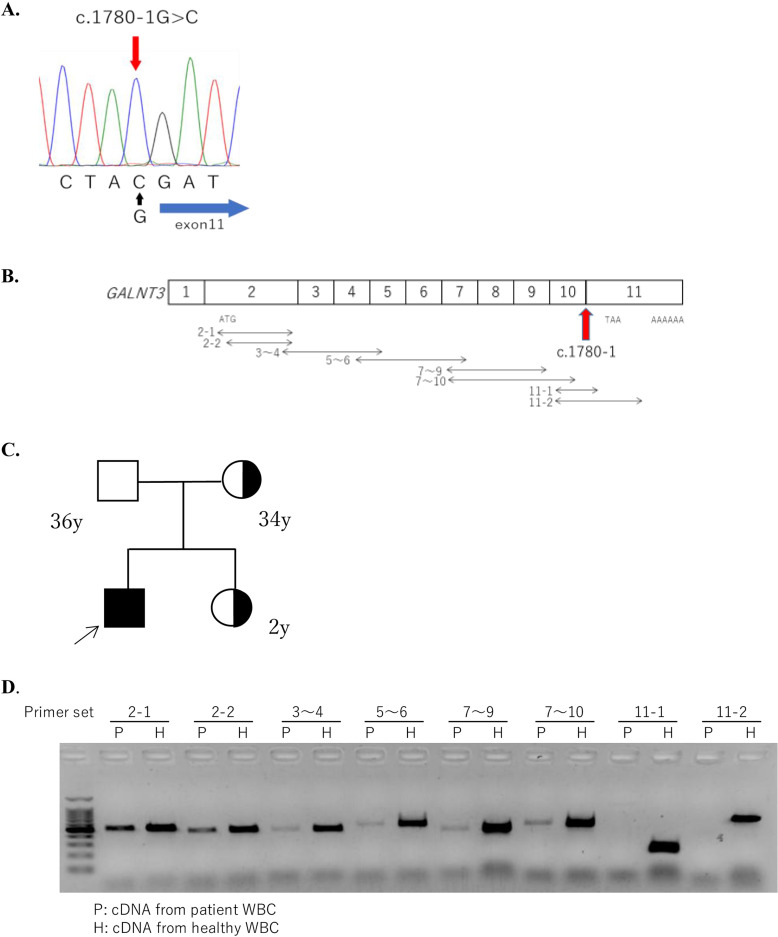
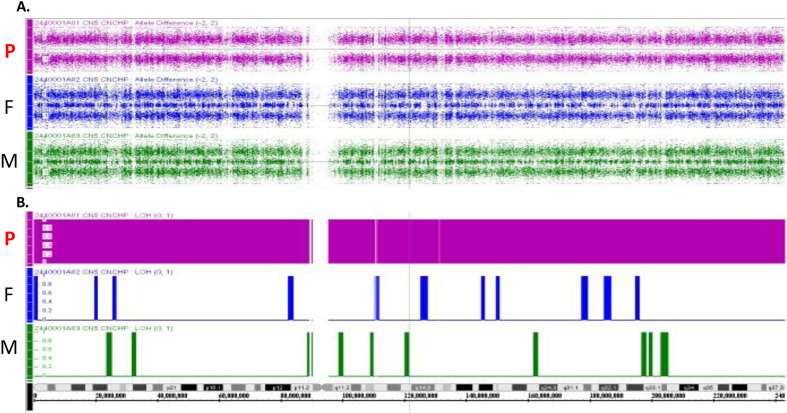
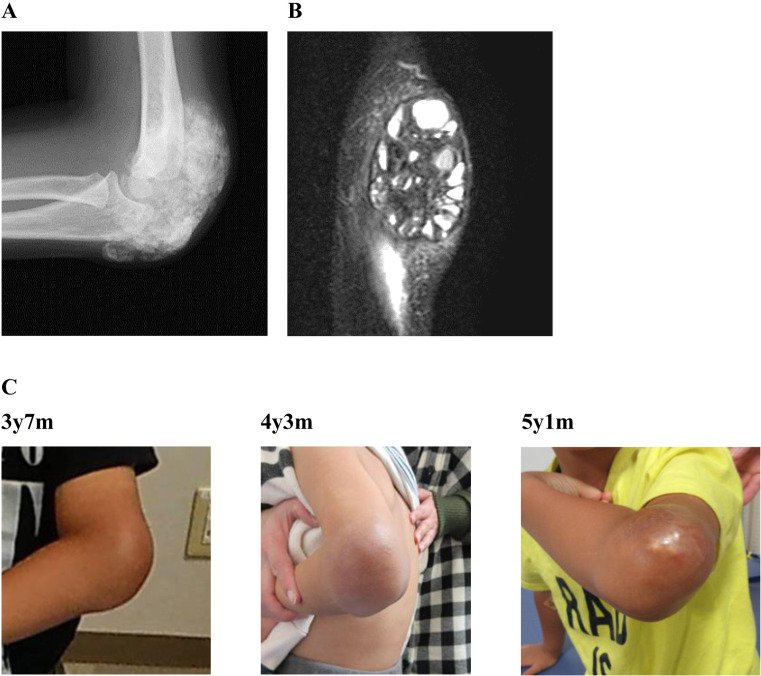
Hyperphosphatemic familial tumoral calcinosis (HFTC) is a rare, inherited autosomal recessive disorder caused by fibroblast growth factor-23 (FGF23), N-acetylgalactosaminyltransferase 3 (GALNT3), or Klotho (KL) gene variants. Here, we report the case of a Japanese boy who presented with a mass in his left elbow at the age of three. Laboratory test results of the patient revealed normocalcemia (10.3 mg/dL) and hyperphosphatemia (8.7 mg/dL); however, despite hyperphosphatemia, serum intact FGF23 level was low, renal tubular reabsorption of phosphate (TRP) level was inappropriately increased, and 1,25-dihydroxyvitamin D3 (1,25(OH)2D3) level was inappropriately normal. Genetic analysis revealed maternal uniparental disomy (UPD) of chromosome 2, which included a novel GALNT3 variant (c.1780-1G>C). Reverse transcription-polymerase chain reaction (RT-PCR) analysis of GALNT3 mRNA confirmed that this variant resulted in the destruction of exon 11. We resected the mass when the patient was five years old, owing to its gradual enlargement. No relapse or new pathological lesions were observed four years after tumor resection. This is the first case report of a Japanese patient with HFTC associated with a novel GALNT3 variant, as well as the first case of HFTC caused by maternal UPD of chromosome 2 that includes the GALNT3 variant.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
