Mahmoudreza Ashrafi, Reyhaneh Kameli, Sareh Hosseinpour, Ehsan Razmara, Zahra Zamani, Zahra Rezaei, Raziyeh Mashayekhi, Neda Pak, Mohammad Barzegar, Reza Azizimalamiri, Morteza Rezvani Kashani, Nahideh Khosroshahi, Maryam Rasulinezhad, Morteza Heidari, Man Amanat, Alireza Abdi, Bahram Mohammadi, Mahmoud Mohammadi, Gholam Reza Zamani, Reza Shervin Badv, Abdolmajid Omrani, Sedigheh Nikbakht, Ali Hosseini Bereshneh, Mojtaba Movahedinia, Hossein Farshad Moghaddam, Hossein Shojaaldini Ardakani, Masood Ghahvechi Akbari, Mehran Beiraghi Tousi, Mohammad Vafaee Shahi, Firouzeh Hosseini, Masoud Hassanvand Amouzadeh, Seyed Ahmad Hosseini, Ali Nikkhah, Ali Khajeh, Hooman Alizadeh, Bahram Yarali, Mohammad Rohani, Parviz Karimi, Hadi Montazer Lotf Elahi, Seyyed Mohamad Mahdi Hosseiny, Masoumeh Sadat Sadeghzadeh, Hossein Mohebbi, Maryam Hosseini Moghadam, Hajar Aryan, Hassan Vahidnezhad, Mahdieh Soveizi, Bahareh Rabbani, Ali Rabbani, Nejat Mahdieh, Masoud Garshasbi, Ali Reza Tavasoli
{"title":"伊朗儿童白细胞营养不良的高度遗传异质性:伊朗白细胞营养障碍登记处的首次报告。","authors":"Mahmoudreza Ashrafi, Reyhaneh Kameli, Sareh Hosseinpour, Ehsan Razmara, Zahra Zamani, Zahra Rezaei, Raziyeh Mashayekhi, Neda Pak, Mohammad Barzegar, Reza Azizimalamiri, Morteza Rezvani Kashani, Nahideh Khosroshahi, Maryam Rasulinezhad, Morteza Heidari, Man Amanat, Alireza Abdi, Bahram Mohammadi, Mahmoud Mohammadi, Gholam Reza Zamani, Reza Shervin Badv, Abdolmajid Omrani, Sedigheh Nikbakht, Ali Hosseini Bereshneh, Mojtaba Movahedinia, Hossein Farshad Moghaddam, Hossein Shojaaldini Ardakani, Masood Ghahvechi Akbari, Mehran Beiraghi Tousi, Mohammad Vafaee Shahi, Firouzeh Hosseini, Masoud Hassanvand Amouzadeh, Seyed Ahmad Hosseini, Ali Nikkhah, Ali Khajeh, Hooman Alizadeh, Bahram Yarali, Mohammad Rohani, Parviz Karimi, Hadi Montazer Lotf Elahi, Seyyed Mohamad Mahdi Hosseiny, Masoumeh Sadat Sadeghzadeh, Hossein Mohebbi, Maryam Hosseini Moghadam, Hajar Aryan, Hassan Vahidnezhad, Mahdieh Soveizi, Bahareh Rabbani, Ali Rabbani, Nejat Mahdieh, Masoud Garshasbi, Ali Reza Tavasoli","doi":"10.1007/s10048-023-00730-y","DOIUrl":null,"url":null,"abstract":"<p><p>Leukodystrophies (LDs) are a heterogeneous group of progressive neurological disorders and characterized by primary involvement of white matter of the central nervous system (CNS). This is the first report of the Iranian LD Registry database to describe the clinical, radiological, and genomic data of Persian patients with leukodystrophies. From 2016 to 2019, patients suspicious of LDs were examined followed by a brain magnetic resonance imaging (MRI). A single gene testing or whole-exome sequencing (WES) was used depending on the neuroradiologic phenotypes. In a few cases, the diagnosis was made by metabolic studies. Based on the MRI pattern, diagnosed patients were divided into cohorts A (hypomyelinating LDs) versus cohort B (Other LDs). The most recent LD classification was utilized for classification of diagnosed patients. For novel variants, in silico analyses were performed to verify their pathogenicity. Out of 680 registered patients, 342 completed the diagnostic evaluations. In total, 245 patients met a diagnosis which in turn 24.5% were categorized in cohort A and the remaining in cohort B. Genetic tests revealed causal variants in 228 patients consisting of 213 variants in 110 genes with 78 novel variants. WES and single gene testing identified a causal variant in 65.5% and 34.5% cases, respectively. The total diagnostic rate of WES was 60.7%. Lysosomal disorders (27.3%; GM2-gangliosidosis-9.8%, MLD-6.1%, KD-4.5%), amino and organic acid disorders (17.15%; Canavan disease-4.5%, L-2-HGA-3.6%), mitochondrial leukodystrophies (12.6%), ion and water homeostasis disorders (7.3%; MLC-4.5%), peroxisomal disorders (6.5%; X-ALD-3.6%), and myelin protein disorders (3.6%; PMLD-3.6%) were the most commonly diagnosed disorders. Thirty-seven percent of cases had a pathogenic variant in nine genes (ARSA, HEXA, ASPA, MLC1, GALC, GJC2, ABCD1, L2HGDH, GCDH). This study highlights the most common types as well as the genetic heterogeneity of LDs in Iranian children.</p>","PeriodicalId":56106,"journal":{"name":"Neurogenetics","volume":" ","pages":"279-289"},"PeriodicalIF":1.2000,"publicationDate":"2023-10-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"High genetic heterogeneity of leukodystrophies in Iranian children: the first report of Iranian Leukodystrophy Registry.\",\"authors\":\"Mahmoudreza Ashrafi, Reyhaneh Kameli, Sareh Hosseinpour, Ehsan Razmara, Zahra Zamani, Zahra Rezaei, Raziyeh Mashayekhi, Neda Pak, Mohammad Barzegar, Reza Azizimalamiri, Morteza Rezvani Kashani, Nahideh Khosroshahi, Maryam Rasulinezhad, Morteza Heidari, Man Amanat, Alireza Abdi, Bahram Mohammadi, Mahmoud Mohammadi, Gholam Reza Zamani, Reza Shervin Badv, Abdolmajid Omrani, Sedigheh Nikbakht, Ali Hosseini Bereshneh, Mojtaba Movahedinia, Hossein Farshad Moghaddam, Hossein Shojaaldini Ardakani, Masood Ghahvechi Akbari, Mehran Beiraghi Tousi, Mohammad Vafaee Shahi, Firouzeh Hosseini, Masoud Hassanvand Amouzadeh, Seyed Ahmad Hosseini, Ali Nikkhah, Ali Khajeh, Hooman Alizadeh, Bahram Yarali, Mohammad Rohani, Parviz Karimi, Hadi Montazer Lotf Elahi, Seyyed Mohamad Mahdi Hosseiny, Masoumeh Sadat Sadeghzadeh, Hossein Mohebbi, Maryam Hosseini Moghadam, Hajar Aryan, Hassan Vahidnezhad, Mahdieh Soveizi, Bahareh Rabbani, Ali Rabbani, Nejat Mahdieh, Masoud Garshasbi, Ali Reza Tavasoli\",\"doi\":\"10.1007/s10048-023-00730-y\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Leukodystrophies (LDs) are a heterogeneous group of progressive neurological disorders and characterized by primary involvement of white matter of the central nervous system (CNS). This is the first report of the Iranian LD Registry database to describe the clinical, radiological, and genomic data of Persian patients with leukodystrophies. From 2016 to 2019, patients suspicious of LDs were examined followed by a brain magnetic resonance imaging (MRI). A single gene testing or whole-exome sequencing (WES) was used depending on the neuroradiologic phenotypes. In a few cases, the diagnosis was made by metabolic studies. Based on the MRI pattern, diagnosed patients were divided into cohorts A (hypomyelinating LDs) versus cohort B (Other LDs). The most recent LD classification was utilized for classification of diagnosed patients. For novel variants, in silico analyses were performed to verify their pathogenicity. Out of 680 registered patients, 342 completed the diagnostic evaluations. In total, 245 patients met a diagnosis which in turn 24.5% were categorized in cohort A and the remaining in cohort B. Genetic tests revealed causal variants in 228 patients consisting of 213 variants in 110 genes with 78 novel variants. WES and single gene testing identified a causal variant in 65.5% and 34.5% cases, respectively. The total diagnostic rate of WES was 60.7%. Lysosomal disorders (27.3%; GM2-gangliosidosis-9.8%, MLD-6.1%, KD-4.5%), amino and organic acid disorders (17.15%; Canavan disease-4.5%, L-2-HGA-3.6%), mitochondrial leukodystrophies (12.6%), ion and water homeostasis disorders (7.3%; MLC-4.5%), peroxisomal disorders (6.5%; X-ALD-3.6%), and myelin protein disorders (3.6%; PMLD-3.6%) were the most commonly diagnosed disorders. Thirty-seven percent of cases had a pathogenic variant in nine genes (ARSA, HEXA, ASPA, MLC1, GALC, GJC2, ABCD1, L2HGDH, GCDH). This study highlights the most common types as well as the genetic heterogeneity of LDs in Iranian children.</p>\",\"PeriodicalId\":56106,\"journal\":{\"name\":\"Neurogenetics\",\"volume\":\" \",\"pages\":\"279-289\"},\"PeriodicalIF\":1.2000,\"publicationDate\":\"2023-10-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Neurogenetics\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://doi.org/10.1007/s10048-023-00730-y\",\"RegionNum\":4,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2023/8/19 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"Q3\",\"JCRName\":\"CLINICAL NEUROLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Neurogenetics","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1007/s10048-023-00730-y","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2023/8/19 0:00:00","PubModel":"Epub","JCR":"Q3","JCRName":"CLINICAL NEUROLOGY","Score":null,"Total":0}
High genetic heterogeneity of leukodystrophies in Iranian children: the first report of Iranian Leukodystrophy Registry.
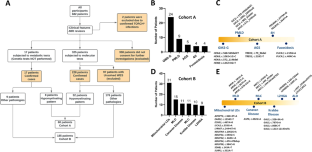
Leukodystrophies (LDs) are a heterogeneous group of progressive neurological disorders and characterized by primary involvement of white matter of the central nervous system (CNS). This is the first report of the Iranian LD Registry database to describe the clinical, radiological, and genomic data of Persian patients with leukodystrophies. From 2016 to 2019, patients suspicious of LDs were examined followed by a brain magnetic resonance imaging (MRI). A single gene testing or whole-exome sequencing (WES) was used depending on the neuroradiologic phenotypes. In a few cases, the diagnosis was made by metabolic studies. Based on the MRI pattern, diagnosed patients were divided into cohorts A (hypomyelinating LDs) versus cohort B (Other LDs). The most recent LD classification was utilized for classification of diagnosed patients. For novel variants, in silico analyses were performed to verify their pathogenicity. Out of 680 registered patients, 342 completed the diagnostic evaluations. In total, 245 patients met a diagnosis which in turn 24.5% were categorized in cohort A and the remaining in cohort B. Genetic tests revealed causal variants in 228 patients consisting of 213 variants in 110 genes with 78 novel variants. WES and single gene testing identified a causal variant in 65.5% and 34.5% cases, respectively. The total diagnostic rate of WES was 60.7%. Lysosomal disorders (27.3%; GM2-gangliosidosis-9.8%, MLD-6.1%, KD-4.5%), amino and organic acid disorders (17.15%; Canavan disease-4.5%, L-2-HGA-3.6%), mitochondrial leukodystrophies (12.6%), ion and water homeostasis disorders (7.3%; MLC-4.5%), peroxisomal disorders (6.5%; X-ALD-3.6%), and myelin protein disorders (3.6%; PMLD-3.6%) were the most commonly diagnosed disorders. Thirty-seven percent of cases had a pathogenic variant in nine genes (ARSA, HEXA, ASPA, MLC1, GALC, GJC2, ABCD1, L2HGDH, GCDH). This study highlights the most common types as well as the genetic heterogeneity of LDs in Iranian children.
期刊介绍:
Neurogenetics publishes findings that contribute to a better understanding of the genetic basis of normal and abnormal function of the nervous system. Neurogenetic disorders are the main focus of the journal. Neurogenetics therefore includes findings in humans and other organisms that help understand neurological disease mechanisms and publishes papers from many different fields such as biophysics, cell biology, human genetics, neuroanatomy, neurochemistry, neurology, neuropathology, neurosurgery and psychiatry.
All papers submitted to Neurogenetics should be of sufficient immediate importance to justify urgent publication. They should present new scientific results. Data merely confirming previously published findings are not acceptable.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
