Frederike L Harms, Deike Weiss, Jasmin Lisfeld, Malik Alawi, Kerstin Kutsche
{"title":"发育性和癫痫性脑病患者的DNM1中的深层内含子变体产生剪接受体位点,仅影响包括外显子10a在内的转录物变体。","authors":"Frederike L Harms, Deike Weiss, Jasmin Lisfeld, Malik Alawi, Kerstin Kutsche","doi":"10.1007/s10048-023-00716-w","DOIUrl":null,"url":null,"abstract":"<p><p>DNM1 developmental and epileptic encephalopathy (DEE) is characterized by severe to profound intellectual disability, hypotonia, movement disorder, and refractory epilepsy, typically presenting with infantile spasms. Most of the affected individuals had de novo missense variants in DNM1. DNM1 undergoes alternative splicing that results in expression of six different transcript variants. One alternatively spliced region affects the tandemly arranged exons 10a and 10b, producing isoforms DNM1A and DNM1B, respectively. Pathogenic variants in the DNM1 coding region affect all transcript variants. Recently, a de novo DNM1 NM_001288739.1:c.1197-8G > A variant located in intron 9 has been reported in several unrelated individuals with DEE that causes in-frame insertion of two amino acids and leads to disease through a dominant-negative mechanism. We report on a patient with DEE and a de novo DNM1 variant NM_001288739.2:c.1197-46C > G in intron 9, upstream of exon 10a. By RT-PCR and Sanger sequencing using fibroblast-derived cDNA of the patient, we identified aberrantly spliced DNM1 mRNAs with exon 9 spliced to the last 45 nucleotides of intron 9 followed by exon 10a (NM_001288739.2:r.1196_1197ins[1197-1_1197-45]). The encoded DNM1A mutant is predicted to contain 15 novel amino acids between Ile398 and Arg399 [NP_001275668.1:p.(Ile398_Arg399ins15)] and likely functions in a dominant-negative manner, similar to other DNM1 mutants. Our data confirm the importance of the DNM1 isoform A for normal human brain function that is underscored by previously reported predominant expression of DMN1A transcripts in pediatric brain, functional differences of the mouse Dnm1a and Dnm1b isoforms, and the Dnm1 fitful mouse, an epilepsy mouse model.</p>","PeriodicalId":56106,"journal":{"name":"Neurogenetics","volume":null,"pages":null},"PeriodicalIF":1.6000,"publicationDate":"2023-07-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"A deep intronic variant in DNM1 in a patient with developmental and epileptic encephalopathy creates a splice acceptor site and affects only transcript variants including exon 10a.\",\"authors\":\"Frederike L Harms, Deike Weiss, Jasmin Lisfeld, Malik Alawi, Kerstin Kutsche\",\"doi\":\"10.1007/s10048-023-00716-w\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>DNM1 developmental and epileptic encephalopathy (DEE) is characterized by severe to profound intellectual disability, hypotonia, movement disorder, and refractory epilepsy, typically presenting with infantile spasms. Most of the affected individuals had de novo missense variants in DNM1. DNM1 undergoes alternative splicing that results in expression of six different transcript variants. One alternatively spliced region affects the tandemly arranged exons 10a and 10b, producing isoforms DNM1A and DNM1B, respectively. Pathogenic variants in the DNM1 coding region affect all transcript variants. Recently, a de novo DNM1 NM_001288739.1:c.1197-8G > A variant located in intron 9 has been reported in several unrelated individuals with DEE that causes in-frame insertion of two amino acids and leads to disease through a dominant-negative mechanism. We report on a patient with DEE and a de novo DNM1 variant NM_001288739.2:c.1197-46C > G in intron 9, upstream of exon 10a. By RT-PCR and Sanger sequencing using fibroblast-derived cDNA of the patient, we identified aberrantly spliced DNM1 mRNAs with exon 9 spliced to the last 45 nucleotides of intron 9 followed by exon 10a (NM_001288739.2:r.1196_1197ins[1197-1_1197-45]). The encoded DNM1A mutant is predicted to contain 15 novel amino acids between Ile398 and Arg399 [NP_001275668.1:p.(Ile398_Arg399ins15)] and likely functions in a dominant-negative manner, similar to other DNM1 mutants. Our data confirm the importance of the DNM1 isoform A for normal human brain function that is underscored by previously reported predominant expression of DMN1A transcripts in pediatric brain, functional differences of the mouse Dnm1a and Dnm1b isoforms, and the Dnm1 fitful mouse, an epilepsy mouse model.</p>\",\"PeriodicalId\":56106,\"journal\":{\"name\":\"Neurogenetics\",\"volume\":null,\"pages\":null},\"PeriodicalIF\":1.6000,\"publicationDate\":\"2023-07-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Neurogenetics\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://doi.org/10.1007/s10048-023-00716-w\",\"RegionNum\":4,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2023/4/11 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"Q3\",\"JCRName\":\"CLINICAL NEUROLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Neurogenetics","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1007/s10048-023-00716-w","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2023/4/11 0:00:00","PubModel":"Epub","JCR":"Q3","JCRName":"CLINICAL NEUROLOGY","Score":null,"Total":0}
A deep intronic variant in DNM1 in a patient with developmental and epileptic encephalopathy creates a splice acceptor site and affects only transcript variants including exon 10a.
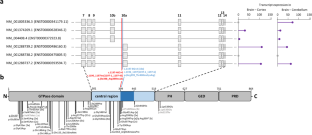
DNM1 developmental and epileptic encephalopathy (DEE) is characterized by severe to profound intellectual disability, hypotonia, movement disorder, and refractory epilepsy, typically presenting with infantile spasms. Most of the affected individuals had de novo missense variants in DNM1. DNM1 undergoes alternative splicing that results in expression of six different transcript variants. One alternatively spliced region affects the tandemly arranged exons 10a and 10b, producing isoforms DNM1A and DNM1B, respectively. Pathogenic variants in the DNM1 coding region affect all transcript variants. Recently, a de novo DNM1 NM_001288739.1:c.1197-8G > A variant located in intron 9 has been reported in several unrelated individuals with DEE that causes in-frame insertion of two amino acids and leads to disease through a dominant-negative mechanism. We report on a patient with DEE and a de novo DNM1 variant NM_001288739.2:c.1197-46C > G in intron 9, upstream of exon 10a. By RT-PCR and Sanger sequencing using fibroblast-derived cDNA of the patient, we identified aberrantly spliced DNM1 mRNAs with exon 9 spliced to the last 45 nucleotides of intron 9 followed by exon 10a (NM_001288739.2:r.1196_1197ins[1197-1_1197-45]). The encoded DNM1A mutant is predicted to contain 15 novel amino acids between Ile398 and Arg399 [NP_001275668.1:p.(Ile398_Arg399ins15)] and likely functions in a dominant-negative manner, similar to other DNM1 mutants. Our data confirm the importance of the DNM1 isoform A for normal human brain function that is underscored by previously reported predominant expression of DMN1A transcripts in pediatric brain, functional differences of the mouse Dnm1a and Dnm1b isoforms, and the Dnm1 fitful mouse, an epilepsy mouse model.
期刊介绍:
Neurogenetics publishes findings that contribute to a better understanding of the genetic basis of normal and abnormal function of the nervous system. Neurogenetic disorders are the main focus of the journal. Neurogenetics therefore includes findings in humans and other organisms that help understand neurological disease mechanisms and publishes papers from many different fields such as biophysics, cell biology, human genetics, neuroanatomy, neurochemistry, neurology, neuropathology, neurosurgery and psychiatry.
All papers submitted to Neurogenetics should be of sufficient immediate importance to justify urgent publication. They should present new scientific results. Data merely confirming previously published findings are not acceptable.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
