Lucy Granat, Debbra Y Knorr, Daniel C Ranson, Emma L Hamer, Ram Prosad Chakrabarty, Francesca Mattedi, Laura Fort-Aznar, Frank Hirth, Sean T Sweeney, Alessio Vagnoni, Navdeep S Chandel, Joseph M Bateman
{"title":"酵母 NDI1 在线粒体复合体 I 缺乏症中重新配置神经元代谢并防止折叠蛋白反应。","authors":"Lucy Granat, Debbra Y Knorr, Daniel C Ranson, Emma L Hamer, Ram Prosad Chakrabarty, Francesca Mattedi, Laura Fort-Aznar, Frank Hirth, Sean T Sweeney, Alessio Vagnoni, Navdeep S Chandel, Joseph M Bateman","doi":"10.1371/journal.pgen.1010793","DOIUrl":null,"url":null,"abstract":"<p><p>Mutations in subunits of the mitochondrial NADH dehydrogenase cause mitochondrial complex I deficiency, a group of severe neurological diseases that can result in death in infancy. The pathogenesis of complex I deficiency remain poorly understood, and as a result there are currently no available treatments. To better understand the underlying mechanisms, we modelled complex I deficiency in Drosophila using knockdown of the mitochondrial complex I subunit ND-75 (NDUFS1) specifically in neurons. Neuronal complex I deficiency causes locomotor defects, seizures and reduced lifespan. At the cellular level, complex I deficiency does not affect ATP levels but leads to mitochondrial morphology defects, reduced endoplasmic reticulum-mitochondria contacts and activation of the endoplasmic reticulum unfolded protein response (UPR) in neurons. Multi-omic analysis shows that complex I deficiency dramatically perturbs mitochondrial metabolism in the brain. We find that expression of the yeast non-proton translocating NADH dehydrogenase NDI1, which reinstates mitochondrial NADH oxidation but not ATP production, restores levels of several key metabolites in the brain in complex I deficiency. Remarkably, NDI1 expression also reinstates endoplasmic reticulum-mitochondria contacts, prevents UPR activation and rescues the behavioural and lifespan phenotypes caused by complex I deficiency. Together, these data show that metabolic disruption due to loss of neuronal NADH dehydrogenase activity cause UPR activation and drive pathogenesis in complex I deficiency.</p>","PeriodicalId":20266,"journal":{"name":"PLoS Genetics","volume":"19 7","pages":"e1010793"},"PeriodicalIF":3.7000,"publicationDate":"2023-07-03","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10348588/pdf/","citationCount":"0","resultStr":"{\"title\":\"Yeast NDI1 reconfigures neuronal metabolism and prevents the unfolded protein response in mitochondrial complex I deficiency.\",\"authors\":\"Lucy Granat, Debbra Y Knorr, Daniel C Ranson, Emma L Hamer, Ram Prosad Chakrabarty, Francesca Mattedi, Laura Fort-Aznar, Frank Hirth, Sean T Sweeney, Alessio Vagnoni, Navdeep S Chandel, Joseph M Bateman\",\"doi\":\"10.1371/journal.pgen.1010793\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Mutations in subunits of the mitochondrial NADH dehydrogenase cause mitochondrial complex I deficiency, a group of severe neurological diseases that can result in death in infancy. The pathogenesis of complex I deficiency remain poorly understood, and as a result there are currently no available treatments. To better understand the underlying mechanisms, we modelled complex I deficiency in Drosophila using knockdown of the mitochondrial complex I subunit ND-75 (NDUFS1) specifically in neurons. Neuronal complex I deficiency causes locomotor defects, seizures and reduced lifespan. At the cellular level, complex I deficiency does not affect ATP levels but leads to mitochondrial morphology defects, reduced endoplasmic reticulum-mitochondria contacts and activation of the endoplasmic reticulum unfolded protein response (UPR) in neurons. Multi-omic analysis shows that complex I deficiency dramatically perturbs mitochondrial metabolism in the brain. We find that expression of the yeast non-proton translocating NADH dehydrogenase NDI1, which reinstates mitochondrial NADH oxidation but not ATP production, restores levels of several key metabolites in the brain in complex I deficiency. Remarkably, NDI1 expression also reinstates endoplasmic reticulum-mitochondria contacts, prevents UPR activation and rescues the behavioural and lifespan phenotypes caused by complex I deficiency. Together, these data show that metabolic disruption due to loss of neuronal NADH dehydrogenase activity cause UPR activation and drive pathogenesis in complex I deficiency.</p>\",\"PeriodicalId\":20266,\"journal\":{\"name\":\"PLoS Genetics\",\"volume\":\"19 7\",\"pages\":\"e1010793\"},\"PeriodicalIF\":3.7000,\"publicationDate\":\"2023-07-03\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10348588/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"PLoS Genetics\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://doi.org/10.1371/journal.pgen.1010793\",\"RegionNum\":2,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2023/7/1 0:00:00\",\"PubModel\":\"eCollection\",\"JCR\":\"Q1\",\"JCRName\":\"Agricultural and Biological Sciences\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"PLoS Genetics","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1371/journal.pgen.1010793","RegionNum":2,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2023/7/1 0:00:00","PubModel":"eCollection","JCR":"Q1","JCRName":"Agricultural and Biological Sciences","Score":null,"Total":0}
引用次数: 0
摘要
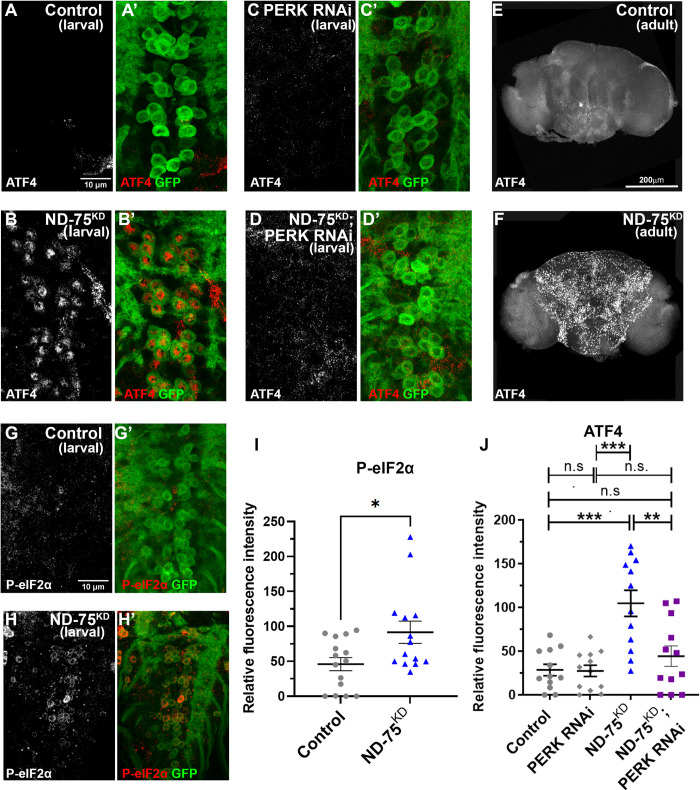
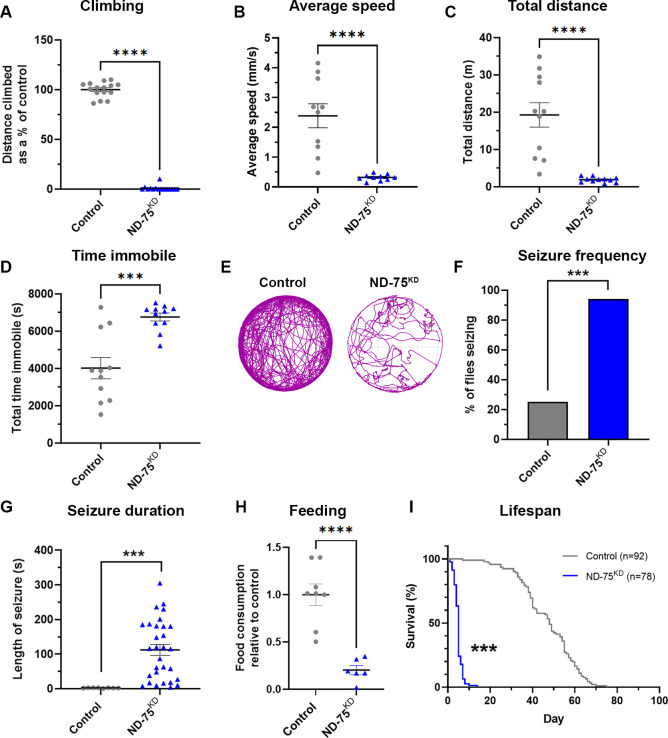
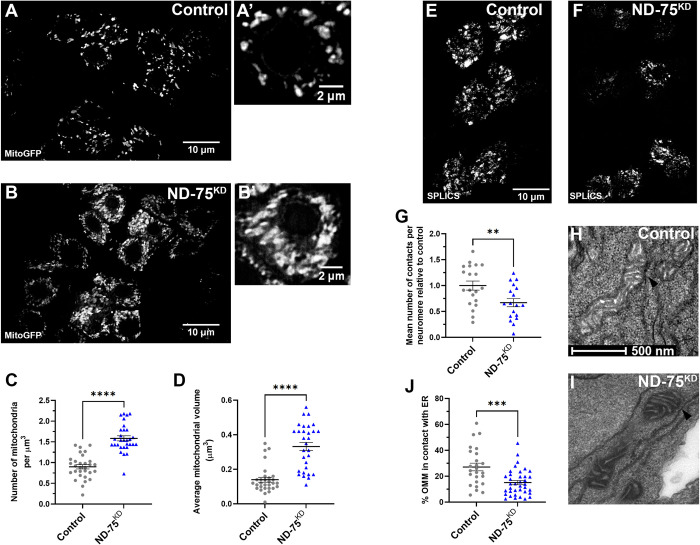
线粒体 NADH 脱氢酶亚基发生突变会导致线粒体复合体 I 缺乏症,这是一组严重的神经系统疾病,可导致婴儿死亡。人们对复合体 I 缺乏症的发病机制仍然知之甚少,因此目前还没有可用的治疗方法。为了更好地了解其潜在机制,我们利用特异性敲除神经元中的线粒体复合体 I 亚基 ND-75 (NDUFS1)来模拟果蝇的复合体 I 缺乏症。神经元复合体 I 缺乏症会导致运动缺陷、癫痫发作和寿命缩短。在细胞水平,复合体 I 缺乏不会影响 ATP 水平,但会导致线粒体形态缺陷、内质网-线粒体接触减少以及激活神经元内质网未折叠蛋白反应(UPR)。多组学分析表明,复合体 I 缺乏会极大地扰乱大脑中的线粒体代谢。我们发现,酵母非质子转运 NADH 脱氢酶 NDI1 能恢复线粒体 NADH 氧化,但不能产生 ATP。值得注意的是,NDI1 的表达还能恢复内质网与线粒体之间的联系,防止 UPR 激活,并挽救复合体 I 缺乏症导致的行为和寿命表型。这些数据共同表明,神经元 NADH 脱氢酶活性丧失导致的代谢紊乱会引起 UPR 激活,并驱动复合体 I 缺乏症的发病机制。
Yeast NDI1 reconfigures neuronal metabolism and prevents the unfolded protein response in mitochondrial complex I deficiency.
Mutations in subunits of the mitochondrial NADH dehydrogenase cause mitochondrial complex I deficiency, a group of severe neurological diseases that can result in death in infancy. The pathogenesis of complex I deficiency remain poorly understood, and as a result there are currently no available treatments. To better understand the underlying mechanisms, we modelled complex I deficiency in Drosophila using knockdown of the mitochondrial complex I subunit ND-75 (NDUFS1) specifically in neurons. Neuronal complex I deficiency causes locomotor defects, seizures and reduced lifespan. At the cellular level, complex I deficiency does not affect ATP levels but leads to mitochondrial morphology defects, reduced endoplasmic reticulum-mitochondria contacts and activation of the endoplasmic reticulum unfolded protein response (UPR) in neurons. Multi-omic analysis shows that complex I deficiency dramatically perturbs mitochondrial metabolism in the brain. We find that expression of the yeast non-proton translocating NADH dehydrogenase NDI1, which reinstates mitochondrial NADH oxidation but not ATP production, restores levels of several key metabolites in the brain in complex I deficiency. Remarkably, NDI1 expression also reinstates endoplasmic reticulum-mitochondria contacts, prevents UPR activation and rescues the behavioural and lifespan phenotypes caused by complex I deficiency. Together, these data show that metabolic disruption due to loss of neuronal NADH dehydrogenase activity cause UPR activation and drive pathogenesis in complex I deficiency.
期刊介绍:
PLOS Genetics is run by an international Editorial Board, headed by the Editors-in-Chief, Greg Barsh (HudsonAlpha Institute of Biotechnology, and Stanford University School of Medicine) and Greg Copenhaver (The University of North Carolina at Chapel Hill).
Articles published in PLOS Genetics are archived in PubMed Central and cited in PubMed.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
