Amal Alotaibi, Veerendra P Gadekar, Pranav Swaroop Gundla, Sumana Mandarthi, Nidhi Jayendra, Asna Tungekar, B V Lavanya, Ashok Kumar Bhagavath, Mary Anne Wong Cordero, Janne Pitkaniemi, Shaik Kalimulla Niazi, Raghavendra Upadhya, Asmatanzeem Bepari, Prashantha Hebbar
{"title":"全球比较转录组揭示新的和群体特异性基因表达在食管鳞状细胞癌。","authors":"Amal Alotaibi, Veerendra P Gadekar, Pranav Swaroop Gundla, Sumana Mandarthi, Nidhi Jayendra, Asna Tungekar, B V Lavanya, Ashok Kumar Bhagavath, Mary Anne Wong Cordero, Janne Pitkaniemi, Shaik Kalimulla Niazi, Raghavendra Upadhya, Asmatanzeem Bepari, Prashantha Hebbar","doi":"10.1186/s13027-023-00525-8","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Esophageal squamous cell carcinoma (ESCC) has a poor prognosis and is one of the deadliest gastrointestinal malignancies. Despite numerous transcriptomics studies to understand its molecular basis, the impact of population-specific differences on this disease remains unexplored.</p><p><strong>Aims: </strong>This study aimed to investigate the population-specific differences in gene expression patterns among ESCC samples obtained from six distinct global populations, identify differentially expressed genes (DEGs) and their associated pathways, and identify potential biomarkers for ESCC diagnosis and prognosis. In addition, this study deciphers population specific microbial and chemical risk factors in ESCC.</p><p><strong>Methods: </strong>We compared the gene expression patterns of ESCC samples from six different global populations by analyzing microarray datasets. To identify DEGs, we conducted stringent quality control and employed linear modeling. We cross-compared the resulting DEG lists of each populations along with ESCC ATLAS to identify known and novel DEGs. We performed a survival analysis using The Cancer Genome Atlas Program (TCGA) data to identify potential biomarkers for ESCC diagnosis and prognosis among the novel DEGs. Finally, we performed comparative functional enrichment and toxicogenomic analysis.</p><p><strong>Results: </strong>Here we report 19 genes with distinct expression patterns among populations, indicating population-specific variations in ESCC. Additionally, we discovered 166 novel DEGs, such as ENDOU, SLCO1B3, KCNS3, IFI35, among others. The survival analysis identified three novel genes (CHRM3, CREG2, H2AC6) critical for ESCC survival. Notably, our findings showed that ECM-related gene ontology terms and pathways were significantly enriched among the DEGs in ESCC. We also found population-specific variations in immune response and microbial infection-related pathways which included genes enriched for HPV, Ameobiosis, Leishmaniosis, and Human Cytomegaloviruses. Our toxicogenomic analysis identified tobacco smoking as the primary risk factor and cisplatin as the main drug chemical interacting with the maximum number of DEGs across populations.</p><p><strong>Conclusion: </strong>This study provides new insights into population-specific differences in gene expression patterns and their associated pathways in ESCC. Our findings suggest that changes in extracellular matrix (ECM) organization may be crucial to the development and progression of this cancer, and that environmental and genetic factors play important roles in the disease. The novel DEGs identified may serve as potential biomarkers for diagnosis, prognosis and treatment.</p>","PeriodicalId":13568,"journal":{"name":"Infectious Agents and Cancer","volume":"18 1","pages":"47"},"PeriodicalIF":2.8000,"publicationDate":"2023-08-28","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10463703/pdf/","citationCount":"1","resultStr":"{\"title\":\"Global comparative transcriptomes uncover novel and population-specific gene expression in esophageal squamous cell carcinoma.\",\"authors\":\"Amal Alotaibi, Veerendra P Gadekar, Pranav Swaroop Gundla, Sumana Mandarthi, Nidhi Jayendra, Asna Tungekar, B V Lavanya, Ashok Kumar Bhagavath, Mary Anne Wong Cordero, Janne Pitkaniemi, Shaik Kalimulla Niazi, Raghavendra Upadhya, Asmatanzeem Bepari, Prashantha Hebbar\",\"doi\":\"10.1186/s13027-023-00525-8\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Background: </strong>Esophageal squamous cell carcinoma (ESCC) has a poor prognosis and is one of the deadliest gastrointestinal malignancies. Despite numerous transcriptomics studies to understand its molecular basis, the impact of population-specific differences on this disease remains unexplored.</p><p><strong>Aims: </strong>This study aimed to investigate the population-specific differences in gene expression patterns among ESCC samples obtained from six distinct global populations, identify differentially expressed genes (DEGs) and their associated pathways, and identify potential biomarkers for ESCC diagnosis and prognosis. In addition, this study deciphers population specific microbial and chemical risk factors in ESCC.</p><p><strong>Methods: </strong>We compared the gene expression patterns of ESCC samples from six different global populations by analyzing microarray datasets. To identify DEGs, we conducted stringent quality control and employed linear modeling. We cross-compared the resulting DEG lists of each populations along with ESCC ATLAS to identify known and novel DEGs. We performed a survival analysis using The Cancer Genome Atlas Program (TCGA) data to identify potential biomarkers for ESCC diagnosis and prognosis among the novel DEGs. Finally, we performed comparative functional enrichment and toxicogenomic analysis.</p><p><strong>Results: </strong>Here we report 19 genes with distinct expression patterns among populations, indicating population-specific variations in ESCC. Additionally, we discovered 166 novel DEGs, such as ENDOU, SLCO1B3, KCNS3, IFI35, among others. The survival analysis identified three novel genes (CHRM3, CREG2, H2AC6) critical for ESCC survival. Notably, our findings showed that ECM-related gene ontology terms and pathways were significantly enriched among the DEGs in ESCC. We also found population-specific variations in immune response and microbial infection-related pathways which included genes enriched for HPV, Ameobiosis, Leishmaniosis, and Human Cytomegaloviruses. Our toxicogenomic analysis identified tobacco smoking as the primary risk factor and cisplatin as the main drug chemical interacting with the maximum number of DEGs across populations.</p><p><strong>Conclusion: </strong>This study provides new insights into population-specific differences in gene expression patterns and their associated pathways in ESCC. Our findings suggest that changes in extracellular matrix (ECM) organization may be crucial to the development and progression of this cancer, and that environmental and genetic factors play important roles in the disease. The novel DEGs identified may serve as potential biomarkers for diagnosis, prognosis and treatment.</p>\",\"PeriodicalId\":13568,\"journal\":{\"name\":\"Infectious Agents and Cancer\",\"volume\":\"18 1\",\"pages\":\"47\"},\"PeriodicalIF\":2.8000,\"publicationDate\":\"2023-08-28\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10463703/pdf/\",\"citationCount\":\"1\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Infectious Agents and Cancer\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://doi.org/10.1186/s13027-023-00525-8\",\"RegionNum\":2,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q3\",\"JCRName\":\"IMMUNOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Infectious Agents and Cancer","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1186/s13027-023-00525-8","RegionNum":2,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"IMMUNOLOGY","Score":null,"Total":0}
Global comparative transcriptomes uncover novel and population-specific gene expression in esophageal squamous cell carcinoma.
Background: Esophageal squamous cell carcinoma (ESCC) has a poor prognosis and is one of the deadliest gastrointestinal malignancies. Despite numerous transcriptomics studies to understand its molecular basis, the impact of population-specific differences on this disease remains unexplored.
Aims: This study aimed to investigate the population-specific differences in gene expression patterns among ESCC samples obtained from six distinct global populations, identify differentially expressed genes (DEGs) and their associated pathways, and identify potential biomarkers for ESCC diagnosis and prognosis. In addition, this study deciphers population specific microbial and chemical risk factors in ESCC.
Methods: We compared the gene expression patterns of ESCC samples from six different global populations by analyzing microarray datasets. To identify DEGs, we conducted stringent quality control and employed linear modeling. We cross-compared the resulting DEG lists of each populations along with ESCC ATLAS to identify known and novel DEGs. We performed a survival analysis using The Cancer Genome Atlas Program (TCGA) data to identify potential biomarkers for ESCC diagnosis and prognosis among the novel DEGs. Finally, we performed comparative functional enrichment and toxicogenomic analysis.
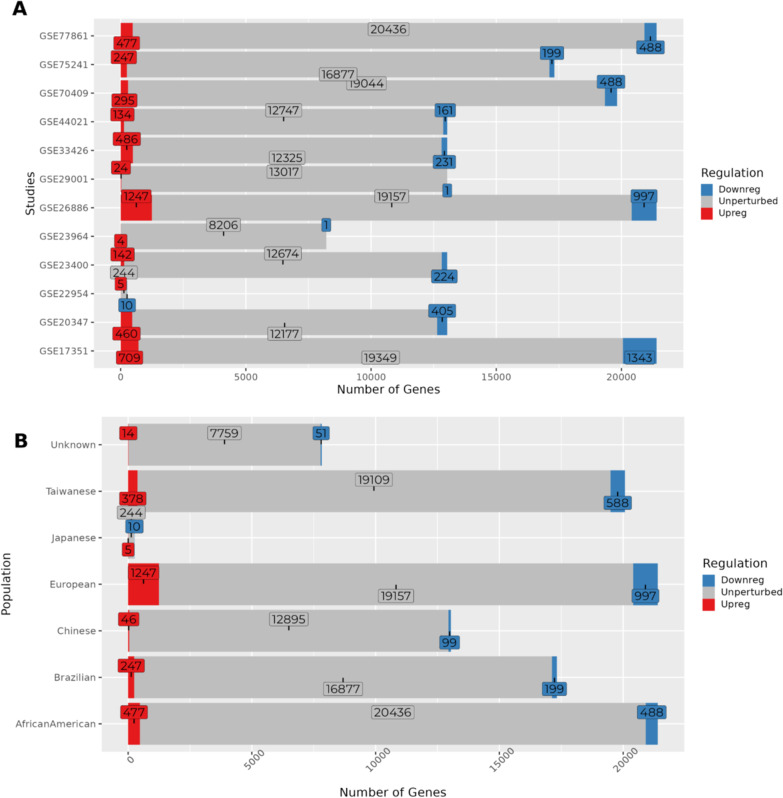
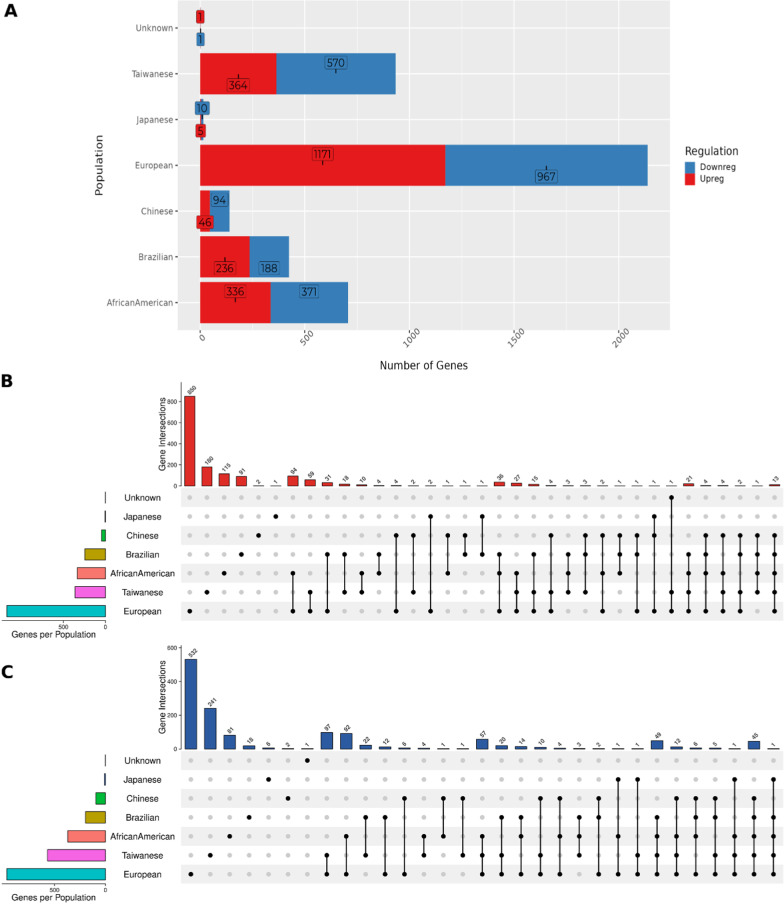
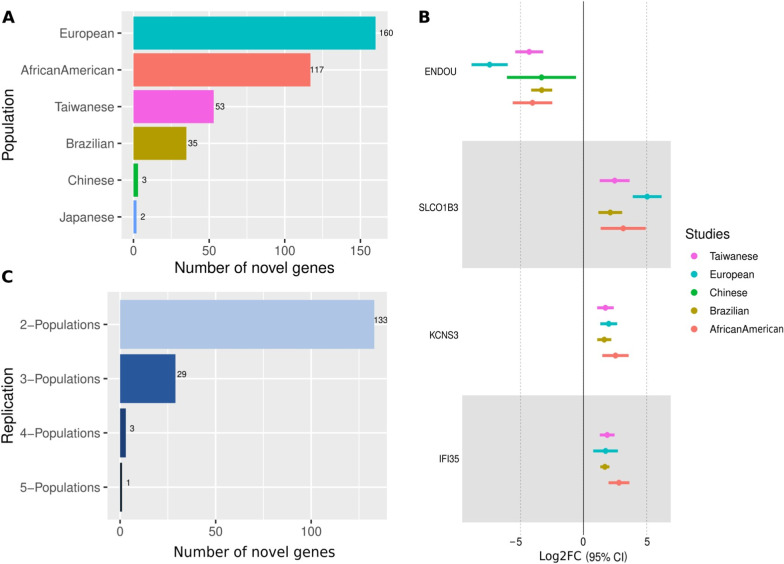
Results: Here we report 19 genes with distinct expression patterns among populations, indicating population-specific variations in ESCC. Additionally, we discovered 166 novel DEGs, such as ENDOU, SLCO1B3, KCNS3, IFI35, among others. The survival analysis identified three novel genes (CHRM3, CREG2, H2AC6) critical for ESCC survival. Notably, our findings showed that ECM-related gene ontology terms and pathways were significantly enriched among the DEGs in ESCC. We also found population-specific variations in immune response and microbial infection-related pathways which included genes enriched for HPV, Ameobiosis, Leishmaniosis, and Human Cytomegaloviruses. Our toxicogenomic analysis identified tobacco smoking as the primary risk factor and cisplatin as the main drug chemical interacting with the maximum number of DEGs across populations.
Conclusion: This study provides new insights into population-specific differences in gene expression patterns and their associated pathways in ESCC. Our findings suggest that changes in extracellular matrix (ECM) organization may be crucial to the development and progression of this cancer, and that environmental and genetic factors play important roles in the disease. The novel DEGs identified may serve as potential biomarkers for diagnosis, prognosis and treatment.
期刊介绍:
Infectious Agents and Cancer is an open access, peer-reviewed online journal that encompasses all aspects of basic, clinical, epidemiological and translational research providing an insight into the association between chronic infections and cancer.
The journal welcomes submissions in the pathogen-related cancer areas and other related topics, in particular:
• HPV and anogenital cancers, as well as head and neck cancers;
• EBV and Burkitt lymphoma;
• HCV/HBV and hepatocellular carcinoma as well as lymphoproliferative diseases;
• HHV8 and Kaposi sarcoma;
• HTLV and leukemia;
• Cancers in Low- and Middle-income countries.
The link between infection and cancer has become well established over the past 50 years, and infection-associated cancer contribute up to 16% of cancers in developed countries and 33% in less developed countries.
Preventive vaccines have been developed for only two cancer-causing viruses, highlighting both the opportunity to prevent infection-associated cancers by vaccination and the gaps that remain before vaccines can be developed for other cancer-causing agents. These gaps are due to incomplete understanding of the basic biology, natural history, epidemiology of many of the pathogens that cause cancer, the mechanisms they exploit to cause cancer, and how to interrupt progression to cancer in human populations. Early diagnosis or identification of lesions at high risk of progression represent the current most critical research area of the field supported by recent advances in genomics and proteomics technologies.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
