Perla Bandini, Nina Borràs, Eugenia Fernandez Mellid, Laura Martin-Fernandez, Paula Melero Valentín, Natalia Comes, Lorena Ramírez, Patricia Cadahia Fernández, Matilde Rodríguez Ruiz, Manuel Mateo Perez Encinas, Francisco Vidal, Irene Corrales
{"title":"首次描述了西班牙由ERCC6L2基因突变引起的骨髓衰竭综合征。","authors":"Perla Bandini, Nina Borràs, Eugenia Fernandez Mellid, Laura Martin-Fernandez, Paula Melero Valentín, Natalia Comes, Lorena Ramírez, Patricia Cadahia Fernández, Matilde Rodríguez Ruiz, Manuel Mateo Perez Encinas, Francisco Vidal, Irene Corrales","doi":"10.1111/bjh.19050","DOIUrl":null,"url":null,"abstract":"<p>Inherited bone marrow failure syndromes (BMFS) are a heterogeneous group of disorders characterized by trilineage peripheral blood cytopenias, often presenting during infancy.<span><sup>1</sup></span> Currently, more than 100 genes, including <i>ERCC6L2</i>, have been associated with BMFS.<span><sup>1</sup></span> <i>ERCC6L2</i> encodes a DNA repair protein of the Snf2 family of helicases called ERCC excision repair 6-like 2 protein. Consequently, variants affecting <i>ERCC6L2</i> cause BMFS-2 and result in genome instability and a predisposition to bone marrow failure (BMF).<span><sup>2</sup></span> In 2014, Tummala et al.<span><sup>2</sup></span> described the first <i>ERCC6L2</i> homozygous variants in two consanguineous families who presented with trilineage BMF with developmental delay and microcephaly.<span><sup>2</sup></span> To our knowledge, only 24 families have been diagnosed with BMFS-2 worldwide and 18 different <i>ERCC6L2</i> variants have been reported (Table 1).<span><sup>2-11</sup></span> Herein, we describe the first case of BMFS-2 in Spain, reporting two siblings with pathogenic variants in <i>ERCC6L2</i> with no extra-haematopoietic symptoms and comparing them with the previously reported patients.</p><p>Two siblings were referred to a haematologist in 2013 at the ages of 13 and 7 years old after a blood test performed for tonsillitis revealed thrombocytopenia and leukopenia in the older brother (33 × 10<sup>9</sup> platelets/L, 3.6 × 10<sup>9</sup> leukocytes/L, 1.3 × 10<sup>9</sup> lymphocytes/L; Figure 1A). The younger brother also presented with thrombocytopenia (82 × 10<sup>9</sup> platelets/L). Based on these findings, a bone marrow (BM) biopsy was conducted for both, detecting BM hypoplasia with no evidence of myelodysplastic syndromes (MDS). Further molecular tests were performed, showing no alterations: karyotype (46, XY), chromosome breakage studies for Fanconi anaemia, and analysis by fluorescence in situ hybridization (FISH) of 20q21, 5q33, 7q35 and the centromeric region of chromosome 8.</p><p>In January 2021 (20 and 14 years old), the siblings were recruited in a research project aimed at unravelling the molecular basis of inherited bleeding disorders or thrombocytopenia by whole-exome sequencing (WES). Written informed consent was provided by their legal guardians, and the study was approved by the research ethics committee of Hospital Universitari Vall d'Hebron in accordance with the guidelines of the Declaration of Helsinki. WES was performed using the DNA Prep with Enrichment protocol (Illumina) in a NextSeq500 system (Illumina). The variants obtained were filtered using a virtual 352-gene panel related to inherited platelet and/or bleeding disorders, but no candidate variants were initially identified.</p><p>In December 2021, the younger brother was admitted to the hospital with fever and upper respiratory infection due to SARS-CoV-2 and severe cytopenia (17 × 10<sup>9</sup> platelets/L, 0.71 × 10<sup>9</sup> neutrophils/L, and 0.5 × 10<sup>9</sup> lymphocytes/L). After recovering from the infection, his blood counts did not improve. Therefore, he was treated with anti-thymocyte globulin, cyclosporine and eltrombopag. Between February and May 2022, he was admitted to the hospital four times for febrile neutropenia with urinary tract infection. On February 2022, a new BM aspirate showed no signs of dysplasia. However, the patient did not respond to the treatment and rapidly progressed to more severe BM aplasia (9 × 10<sup>9</sup> platelets/L, 0.5 × 10<sup>9</sup> leukocytes/L). At that time, reanalysis of the WES filtering within 772 genes associated with BM aplasia enabled the identification of two truncating variants in <i>ERCC6L2</i> in both siblings.</p><p>The <i>ERCC6L2</i> variants, classified as pathogenic according to American College of Medical Genetics guidelines, were: (1) NM_020207:c.1930C>T, p.(Arg644Ter) in exon 13, previously described as c.1963C>T, p.(Arg655Ter), the second most recurrent variant only found in homozygous state and located in the helicase domain;<span><sup>2, 3, 7</sup></span> and (2) NM_020207:c.2156del, p.(Gly719AspfsTer50) in exon 15, previously described as c.2189del, p.(Gly730AspfsTer50) identified in a single patient in heterozygous state (Figure 1B).<span><sup>4</sup></span> Sanger sequencing of exons harbouring the pathogenic variants confirmed its presence in the siblings and the carrier status of non-consanguineous parents for each mutation (Figure 1C).</p><p>Our results show that recognition of patients with BMFS-2 can be challenging, as the initial signs are non-specific cytopenias, and the association with the extra-haematopoietic symptoms may divert the investigation. Notably, for this family, <i>ERCC6L2</i> variants were initially ruled out as the cause of the disease due to the linkage of BMFS-2 with microcephaly in the Online Mendelian Inheritance in Man database (OMIM#615715). According to the studies published to date, extrahaematological involvement is rare (15%) and is mainly observed (83%) in consanguineous families.<span><sup>2-5, 7</sup></span> Consequently, as we present two new patients without extrahaematological features, we question the association between <i>ERCC6L2</i> variants and neurological impairments.</p><p>On June 2022, the younger brother underwent an allogeneic BM transplant due to the disease evolution and lack of response to treatment, and 6 months later his blood counts were restored (163 × 10<sup>9</sup> platelet/L and 4.74 × 10<sup>9</sup> leukocytes/L). In the literature, eight out of 34 patients (24%) belonging to 24 families underwent haematopoietic stem cell transplantation (HSCT). The three patients submitted to HSCT before the development of MDS and/or acute myeloid leukaemia (AML) survived, whereas, for the other five patients, who received HSCT during MDS or AML, the outcomes were unfavourable for four of them and unknown for the last case.<span><sup>12</sup></span> Although the number of cases is too small to establish definitive conclusions, the results seem to point out the critical importance of early diagnosis of BMFS-2 for better prognosis and treatment outcomes. However, more evidence will be needed to support this finding.</p><p>Despite both siblings having the same <i>ERCC6L2</i> variants, the younger brother presented with a more severe phenotype, experiencing sudden progression to severe BM aplasia. Although the BMFS-2 has a high penetrance, Bluteau et al.<span><sup>7</sup></span> also reported the case of two siblings who presented the same <i>ERCC6L2</i> variants but had a very different evolution: one patient had only thrombocytopenia and neutropenia, whereas her sister also developed MDS with chromosome 7 monosomy. This highlights how the pathogenic mechanisms that are triggered in patients with <i>ERCC6L2</i> variants still need to be further elucidated.</p><p>A recent in vitro cellular study<span><sup>13</sup></span> has demonstrated that loss of <i>ERCC6L2</i> results in up-regulation of DNA repair and <i>TP53</i> pathways, leading to a significant impairment of HSC clonogenic potential, delayed erythropoiesis and lineage skewing in the BM. Consequently, the authors suggested that patients with <i>ERCC6L2</i> variants have a microenvironment that is primed for malignancy. <i>TP53</i> mutated clones were described in seven out of the 12 patients (35%) with BMFS-2 who developed MDS/AML, indicating that this acquired alteration may represent the first step toward malignancy.<span><sup>12</sup></span> In this sense, it is noteworthy that somatic mutation analysis in the younger sibling revealed two frameshift variants, one in <i>NPM1</i>: NM_001355006:c.80_863dup, p.(Trp288CysfsTer12); and the other in <i>BCOR</i>: NM_001123385:c.4412_4415dup, p.(Arg1472SerfsTer6), which are known players in AML development.<span><sup>14</sup></span></p><p>In conclusion, we report the first description of BM aplasia caused by <i>ERCC6L2</i> variants in Spain and the fifth family with BMFS-2 caused by compound heterozygous variants in <i>ERCC6L2</i>. Our findings contribute to the small worldwide cohort of 24 families with BMFS-2 and report two more patients without extra-haematological involvement, providing further evidence for the weak association of microcephaly and neurodevelopmental delay with this syndrome. In our study, the younger patient with severe BM aplasia received HSCT before potentially developing AML and had a favourable outcome, resulting in the restoration of BM cell counts. Early detection of the molecular defect through genetic analysis is essential for guiding prognosis, improving family counselling, and patient management, which is especially relevant given the high susceptibility of BMFS-2 patients to the development of MDS and AML.</p><p>Irene Corrales and Francisco Vidal developed the hypothesis and designed the research; Eugenia Fernandez Mellid, Paula Melero Valentín, Patricia Cadahia Fernández, Matilde Rodríguez Ruiz, Manuel Mateo Perez Encinas recruited study participants, collected samples and performed laboratory tests; Perla Bandini, Natalia Comes, and Lorena Ramírez performed WES and conducted analysis of sequencing data; Perla Bandini and Nina Borràs interpreted the data; Perla Bandini, drafted the initial manuscript with help from Nina Borràs, Irene Corrales and Laura Martin-Fernandez; and all authors read and approved the final version of the manuscript.</p><p>This study was supported by the Spanish Ministry of the Economy and Competitiveness (MINECO, Ministerio de Economía y Competitividad), Instituto de Salud Carlos III (ISCIII) (PI18/01492) and Fundació Privada Catalana de l'Hemofília.</p><p>No conflicts of interest declared.</p><p>The study was approved by the Research Ethics Committee of Hospital Universitari Vall d'Hebron in accordance with the guidelines of the Declaration of Helsinki.</p><p>Written informed consent was provided by their legal guardians.</p>","PeriodicalId":135,"journal":{"name":"British Journal of Haematology","volume":"203 4","pages":"e102-e107"},"PeriodicalIF":3.8000,"publicationDate":"2023-09-11","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1111/bjh.19050","citationCount":"0","resultStr":"{\"title\":\"First description of bone marrow failure syndrome in Spain caused by mutations in the ERCC6L2 gene\",\"authors\":\"Perla Bandini, Nina Borràs, Eugenia Fernandez Mellid, Laura Martin-Fernandez, Paula Melero Valentín, Natalia Comes, Lorena Ramírez, Patricia Cadahia Fernández, Matilde Rodríguez Ruiz, Manuel Mateo Perez Encinas, Francisco Vidal, Irene Corrales\",\"doi\":\"10.1111/bjh.19050\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p>Inherited bone marrow failure syndromes (BMFS) are a heterogeneous group of disorders characterized by trilineage peripheral blood cytopenias, often presenting during infancy.<span><sup>1</sup></span> Currently, more than 100 genes, including <i>ERCC6L2</i>, have been associated with BMFS.<span><sup>1</sup></span> <i>ERCC6L2</i> encodes a DNA repair protein of the Snf2 family of helicases called ERCC excision repair 6-like 2 protein. Consequently, variants affecting <i>ERCC6L2</i> cause BMFS-2 and result in genome instability and a predisposition to bone marrow failure (BMF).<span><sup>2</sup></span> In 2014, Tummala et al.<span><sup>2</sup></span> described the first <i>ERCC6L2</i> homozygous variants in two consanguineous families who presented with trilineage BMF with developmental delay and microcephaly.<span><sup>2</sup></span> To our knowledge, only 24 families have been diagnosed with BMFS-2 worldwide and 18 different <i>ERCC6L2</i> variants have been reported (Table 1).<span><sup>2-11</sup></span> Herein, we describe the first case of BMFS-2 in Spain, reporting two siblings with pathogenic variants in <i>ERCC6L2</i> with no extra-haematopoietic symptoms and comparing them with the previously reported patients.</p><p>Two siblings were referred to a haematologist in 2013 at the ages of 13 and 7 years old after a blood test performed for tonsillitis revealed thrombocytopenia and leukopenia in the older brother (33 × 10<sup>9</sup> platelets/L, 3.6 × 10<sup>9</sup> leukocytes/L, 1.3 × 10<sup>9</sup> lymphocytes/L; Figure 1A). The younger brother also presented with thrombocytopenia (82 × 10<sup>9</sup> platelets/L). Based on these findings, a bone marrow (BM) biopsy was conducted for both, detecting BM hypoplasia with no evidence of myelodysplastic syndromes (MDS). Further molecular tests were performed, showing no alterations: karyotype (46, XY), chromosome breakage studies for Fanconi anaemia, and analysis by fluorescence in situ hybridization (FISH) of 20q21, 5q33, 7q35 and the centromeric region of chromosome 8.</p><p>In January 2021 (20 and 14 years old), the siblings were recruited in a research project aimed at unravelling the molecular basis of inherited bleeding disorders or thrombocytopenia by whole-exome sequencing (WES). Written informed consent was provided by their legal guardians, and the study was approved by the research ethics committee of Hospital Universitari Vall d'Hebron in accordance with the guidelines of the Declaration of Helsinki. WES was performed using the DNA Prep with Enrichment protocol (Illumina) in a NextSeq500 system (Illumina). The variants obtained were filtered using a virtual 352-gene panel related to inherited platelet and/or bleeding disorders, but no candidate variants were initially identified.</p><p>In December 2021, the younger brother was admitted to the hospital with fever and upper respiratory infection due to SARS-CoV-2 and severe cytopenia (17 × 10<sup>9</sup> platelets/L, 0.71 × 10<sup>9</sup> neutrophils/L, and 0.5 × 10<sup>9</sup> lymphocytes/L). After recovering from the infection, his blood counts did not improve. Therefore, he was treated with anti-thymocyte globulin, cyclosporine and eltrombopag. Between February and May 2022, he was admitted to the hospital four times for febrile neutropenia with urinary tract infection. On February 2022, a new BM aspirate showed no signs of dysplasia. However, the patient did not respond to the treatment and rapidly progressed to more severe BM aplasia (9 × 10<sup>9</sup> platelets/L, 0.5 × 10<sup>9</sup> leukocytes/L). At that time, reanalysis of the WES filtering within 772 genes associated with BM aplasia enabled the identification of two truncating variants in <i>ERCC6L2</i> in both siblings.</p><p>The <i>ERCC6L2</i> variants, classified as pathogenic according to American College of Medical Genetics guidelines, were: (1) NM_020207:c.1930C>T, p.(Arg644Ter) in exon 13, previously described as c.1963C>T, p.(Arg655Ter), the second most recurrent variant only found in homozygous state and located in the helicase domain;<span><sup>2, 3, 7</sup></span> and (2) NM_020207:c.2156del, p.(Gly719AspfsTer50) in exon 15, previously described as c.2189del, p.(Gly730AspfsTer50) identified in a single patient in heterozygous state (Figure 1B).<span><sup>4</sup></span> Sanger sequencing of exons harbouring the pathogenic variants confirmed its presence in the siblings and the carrier status of non-consanguineous parents for each mutation (Figure 1C).</p><p>Our results show that recognition of patients with BMFS-2 can be challenging, as the initial signs are non-specific cytopenias, and the association with the extra-haematopoietic symptoms may divert the investigation. Notably, for this family, <i>ERCC6L2</i> variants were initially ruled out as the cause of the disease due to the linkage of BMFS-2 with microcephaly in the Online Mendelian Inheritance in Man database (OMIM#615715). According to the studies published to date, extrahaematological involvement is rare (15%) and is mainly observed (83%) in consanguineous families.<span><sup>2-5, 7</sup></span> Consequently, as we present two new patients without extrahaematological features, we question the association between <i>ERCC6L2</i> variants and neurological impairments.</p><p>On June 2022, the younger brother underwent an allogeneic BM transplant due to the disease evolution and lack of response to treatment, and 6 months later his blood counts were restored (163 × 10<sup>9</sup> platelet/L and 4.74 × 10<sup>9</sup> leukocytes/L). In the literature, eight out of 34 patients (24%) belonging to 24 families underwent haematopoietic stem cell transplantation (HSCT). The three patients submitted to HSCT before the development of MDS and/or acute myeloid leukaemia (AML) survived, whereas, for the other five patients, who received HSCT during MDS or AML, the outcomes were unfavourable for four of them and unknown for the last case.<span><sup>12</sup></span> Although the number of cases is too small to establish definitive conclusions, the results seem to point out the critical importance of early diagnosis of BMFS-2 for better prognosis and treatment outcomes. However, more evidence will be needed to support this finding.</p><p>Despite both siblings having the same <i>ERCC6L2</i> variants, the younger brother presented with a more severe phenotype, experiencing sudden progression to severe BM aplasia. Although the BMFS-2 has a high penetrance, Bluteau et al.<span><sup>7</sup></span> also reported the case of two siblings who presented the same <i>ERCC6L2</i> variants but had a very different evolution: one patient had only thrombocytopenia and neutropenia, whereas her sister also developed MDS with chromosome 7 monosomy. This highlights how the pathogenic mechanisms that are triggered in patients with <i>ERCC6L2</i> variants still need to be further elucidated.</p><p>A recent in vitro cellular study<span><sup>13</sup></span> has demonstrated that loss of <i>ERCC6L2</i> results in up-regulation of DNA repair and <i>TP53</i> pathways, leading to a significant impairment of HSC clonogenic potential, delayed erythropoiesis and lineage skewing in the BM. Consequently, the authors suggested that patients with <i>ERCC6L2</i> variants have a microenvironment that is primed for malignancy. <i>TP53</i> mutated clones were described in seven out of the 12 patients (35%) with BMFS-2 who developed MDS/AML, indicating that this acquired alteration may represent the first step toward malignancy.<span><sup>12</sup></span> In this sense, it is noteworthy that somatic mutation analysis in the younger sibling revealed two frameshift variants, one in <i>NPM1</i>: NM_001355006:c.80_863dup, p.(Trp288CysfsTer12); and the other in <i>BCOR</i>: NM_001123385:c.4412_4415dup, p.(Arg1472SerfsTer6), which are known players in AML development.<span><sup>14</sup></span></p><p>In conclusion, we report the first description of BM aplasia caused by <i>ERCC6L2</i> variants in Spain and the fifth family with BMFS-2 caused by compound heterozygous variants in <i>ERCC6L2</i>. Our findings contribute to the small worldwide cohort of 24 families with BMFS-2 and report two more patients without extra-haematological involvement, providing further evidence for the weak association of microcephaly and neurodevelopmental delay with this syndrome. In our study, the younger patient with severe BM aplasia received HSCT before potentially developing AML and had a favourable outcome, resulting in the restoration of BM cell counts. Early detection of the molecular defect through genetic analysis is essential for guiding prognosis, improving family counselling, and patient management, which is especially relevant given the high susceptibility of BMFS-2 patients to the development of MDS and AML.</p><p>Irene Corrales and Francisco Vidal developed the hypothesis and designed the research; Eugenia Fernandez Mellid, Paula Melero Valentín, Patricia Cadahia Fernández, Matilde Rodríguez Ruiz, Manuel Mateo Perez Encinas recruited study participants, collected samples and performed laboratory tests; Perla Bandini, Natalia Comes, and Lorena Ramírez performed WES and conducted analysis of sequencing data; Perla Bandini and Nina Borràs interpreted the data; Perla Bandini, drafted the initial manuscript with help from Nina Borràs, Irene Corrales and Laura Martin-Fernandez; and all authors read and approved the final version of the manuscript.</p><p>This study was supported by the Spanish Ministry of the Economy and Competitiveness (MINECO, Ministerio de Economía y Competitividad), Instituto de Salud Carlos III (ISCIII) (PI18/01492) and Fundació Privada Catalana de l'Hemofília.</p><p>No conflicts of interest declared.</p><p>The study was approved by the Research Ethics Committee of Hospital Universitari Vall d'Hebron in accordance with the guidelines of the Declaration of Helsinki.</p><p>Written informed consent was provided by their legal guardians.</p>\",\"PeriodicalId\":135,\"journal\":{\"name\":\"British Journal of Haematology\",\"volume\":\"203 4\",\"pages\":\"e102-e107\"},\"PeriodicalIF\":3.8000,\"publicationDate\":\"2023-09-11\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://onlinelibrary.wiley.com/doi/epdf/10.1111/bjh.19050\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"British Journal of Haematology\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://onlinelibrary.wiley.com/doi/10.1111/bjh.19050\",\"RegionNum\":2,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"HEMATOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"British Journal of Haematology","FirstCategoryId":"3","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1111/bjh.19050","RegionNum":2,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"HEMATOLOGY","Score":null,"Total":0}
First description of bone marrow failure syndrome in Spain caused by mutations in the ERCC6L2 gene
Inherited bone marrow failure syndromes (BMFS) are a heterogeneous group of disorders characterized by trilineage peripheral blood cytopenias, often presenting during infancy.1 Currently, more than 100 genes, including ERCC6L2, have been associated with BMFS.1ERCC6L2 encodes a DNA repair protein of the Snf2 family of helicases called ERCC excision repair 6-like 2 protein. Consequently, variants affecting ERCC6L2 cause BMFS-2 and result in genome instability and a predisposition to bone marrow failure (BMF).2 In 2014, Tummala et al.2 described the first ERCC6L2 homozygous variants in two consanguineous families who presented with trilineage BMF with developmental delay and microcephaly.2 To our knowledge, only 24 families have been diagnosed with BMFS-2 worldwide and 18 different ERCC6L2 variants have been reported (Table 1).2-11 Herein, we describe the first case of BMFS-2 in Spain, reporting two siblings with pathogenic variants in ERCC6L2 with no extra-haematopoietic symptoms and comparing them with the previously reported patients.
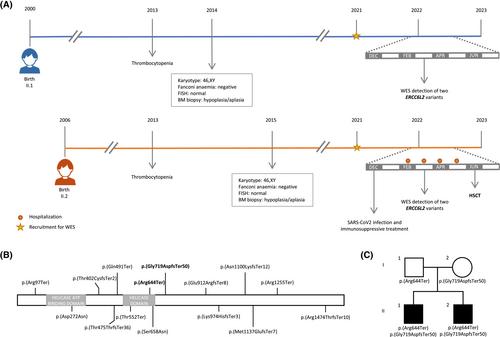
Two siblings were referred to a haematologist in 2013 at the ages of 13 and 7 years old after a blood test performed for tonsillitis revealed thrombocytopenia and leukopenia in the older brother (33 × 109 platelets/L, 3.6 × 109 leukocytes/L, 1.3 × 109 lymphocytes/L; Figure 1A). The younger brother also presented with thrombocytopenia (82 × 109 platelets/L). Based on these findings, a bone marrow (BM) biopsy was conducted for both, detecting BM hypoplasia with no evidence of myelodysplastic syndromes (MDS). Further molecular tests were performed, showing no alterations: karyotype (46, XY), chromosome breakage studies for Fanconi anaemia, and analysis by fluorescence in situ hybridization (FISH) of 20q21, 5q33, 7q35 and the centromeric region of chromosome 8.
In January 2021 (20 and 14 years old), the siblings were recruited in a research project aimed at unravelling the molecular basis of inherited bleeding disorders or thrombocytopenia by whole-exome sequencing (WES). Written informed consent was provided by their legal guardians, and the study was approved by the research ethics committee of Hospital Universitari Vall d'Hebron in accordance with the guidelines of the Declaration of Helsinki. WES was performed using the DNA Prep with Enrichment protocol (Illumina) in a NextSeq500 system (Illumina). The variants obtained were filtered using a virtual 352-gene panel related to inherited platelet and/or bleeding disorders, but no candidate variants were initially identified.
In December 2021, the younger brother was admitted to the hospital with fever and upper respiratory infection due to SARS-CoV-2 and severe cytopenia (17 × 109 platelets/L, 0.71 × 109 neutrophils/L, and 0.5 × 109 lymphocytes/L). After recovering from the infection, his blood counts did not improve. Therefore, he was treated with anti-thymocyte globulin, cyclosporine and eltrombopag. Between February and May 2022, he was admitted to the hospital four times for febrile neutropenia with urinary tract infection. On February 2022, a new BM aspirate showed no signs of dysplasia. However, the patient did not respond to the treatment and rapidly progressed to more severe BM aplasia (9 × 109 platelets/L, 0.5 × 109 leukocytes/L). At that time, reanalysis of the WES filtering within 772 genes associated with BM aplasia enabled the identification of two truncating variants in ERCC6L2 in both siblings.
The ERCC6L2 variants, classified as pathogenic according to American College of Medical Genetics guidelines, were: (1) NM_020207:c.1930C>T, p.(Arg644Ter) in exon 13, previously described as c.1963C>T, p.(Arg655Ter), the second most recurrent variant only found in homozygous state and located in the helicase domain;2, 3, 7 and (2) NM_020207:c.2156del, p.(Gly719AspfsTer50) in exon 15, previously described as c.2189del, p.(Gly730AspfsTer50) identified in a single patient in heterozygous state (Figure 1B).4 Sanger sequencing of exons harbouring the pathogenic variants confirmed its presence in the siblings and the carrier status of non-consanguineous parents for each mutation (Figure 1C).
Our results show that recognition of patients with BMFS-2 can be challenging, as the initial signs are non-specific cytopenias, and the association with the extra-haematopoietic symptoms may divert the investigation. Notably, for this family, ERCC6L2 variants were initially ruled out as the cause of the disease due to the linkage of BMFS-2 with microcephaly in the Online Mendelian Inheritance in Man database (OMIM#615715). According to the studies published to date, extrahaematological involvement is rare (15%) and is mainly observed (83%) in consanguineous families.2-5, 7 Consequently, as we present two new patients without extrahaematological features, we question the association between ERCC6L2 variants and neurological impairments.
On June 2022, the younger brother underwent an allogeneic BM transplant due to the disease evolution and lack of response to treatment, and 6 months later his blood counts were restored (163 × 109 platelet/L and 4.74 × 109 leukocytes/L). In the literature, eight out of 34 patients (24%) belonging to 24 families underwent haematopoietic stem cell transplantation (HSCT). The three patients submitted to HSCT before the development of MDS and/or acute myeloid leukaemia (AML) survived, whereas, for the other five patients, who received HSCT during MDS or AML, the outcomes were unfavourable for four of them and unknown for the last case.12 Although the number of cases is too small to establish definitive conclusions, the results seem to point out the critical importance of early diagnosis of BMFS-2 for better prognosis and treatment outcomes. However, more evidence will be needed to support this finding.
Despite both siblings having the same ERCC6L2 variants, the younger brother presented with a more severe phenotype, experiencing sudden progression to severe BM aplasia. Although the BMFS-2 has a high penetrance, Bluteau et al.7 also reported the case of two siblings who presented the same ERCC6L2 variants but had a very different evolution: one patient had only thrombocytopenia and neutropenia, whereas her sister also developed MDS with chromosome 7 monosomy. This highlights how the pathogenic mechanisms that are triggered in patients with ERCC6L2 variants still need to be further elucidated.
A recent in vitro cellular study13 has demonstrated that loss of ERCC6L2 results in up-regulation of DNA repair and TP53 pathways, leading to a significant impairment of HSC clonogenic potential, delayed erythropoiesis and lineage skewing in the BM. Consequently, the authors suggested that patients with ERCC6L2 variants have a microenvironment that is primed for malignancy. TP53 mutated clones were described in seven out of the 12 patients (35%) with BMFS-2 who developed MDS/AML, indicating that this acquired alteration may represent the first step toward malignancy.12 In this sense, it is noteworthy that somatic mutation analysis in the younger sibling revealed two frameshift variants, one in NPM1: NM_001355006:c.80_863dup, p.(Trp288CysfsTer12); and the other in BCOR: NM_001123385:c.4412_4415dup, p.(Arg1472SerfsTer6), which are known players in AML development.14
In conclusion, we report the first description of BM aplasia caused by ERCC6L2 variants in Spain and the fifth family with BMFS-2 caused by compound heterozygous variants in ERCC6L2. Our findings contribute to the small worldwide cohort of 24 families with BMFS-2 and report two more patients without extra-haematological involvement, providing further evidence for the weak association of microcephaly and neurodevelopmental delay with this syndrome. In our study, the younger patient with severe BM aplasia received HSCT before potentially developing AML and had a favourable outcome, resulting in the restoration of BM cell counts. Early detection of the molecular defect through genetic analysis is essential for guiding prognosis, improving family counselling, and patient management, which is especially relevant given the high susceptibility of BMFS-2 patients to the development of MDS and AML.
Irene Corrales and Francisco Vidal developed the hypothesis and designed the research; Eugenia Fernandez Mellid, Paula Melero Valentín, Patricia Cadahia Fernández, Matilde Rodríguez Ruiz, Manuel Mateo Perez Encinas recruited study participants, collected samples and performed laboratory tests; Perla Bandini, Natalia Comes, and Lorena Ramírez performed WES and conducted analysis of sequencing data; Perla Bandini and Nina Borràs interpreted the data; Perla Bandini, drafted the initial manuscript with help from Nina Borràs, Irene Corrales and Laura Martin-Fernandez; and all authors read and approved the final version of the manuscript.
This study was supported by the Spanish Ministry of the Economy and Competitiveness (MINECO, Ministerio de Economía y Competitividad), Instituto de Salud Carlos III (ISCIII) (PI18/01492) and Fundació Privada Catalana de l'Hemofília.
No conflicts of interest declared.
The study was approved by the Research Ethics Committee of Hospital Universitari Vall d'Hebron in accordance with the guidelines of the Declaration of Helsinki.
Written informed consent was provided by their legal guardians.
期刊介绍:
The British Journal of Haematology publishes original research papers in clinical, laboratory and experimental haematology. The Journal also features annotations, reviews, short reports, images in haematology and Letters to the Editor.



 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
