{"title":"DeepMHCI:一个锚定位置感知的深度相互作用模型,用于准确预测MHC-I肽结合亲和力。","authors":"Wei Qu, Ronghui You, Hiroshi Mamitsuka, Shanfeng Zhu","doi":"10.1093/bioinformatics/btad551","DOIUrl":null,"url":null,"abstract":"<p><strong>Motivation: </strong>Computationally predicting major histocompatibility complex class I (MHC-I) peptide binding affinity is an important problem in immunological bioinformatics, which is also crucial for the identification of neoantigens for personalized therapeutic cancer vaccines. Recent cutting-edge deep learning-based methods for this problem cannot achieve satisfactory performance, especially for non-9-mer peptides. This is because such methods generate the input by simply concatenating the two given sequences: a peptide and (the pseudo sequence of) an MHC class I molecule, which cannot precisely capture the anchor positions of the MHC binding motif for the peptides with variable lengths. We thus developed an anchor position-aware and high-performance deep model, DeepMHCI, with a position-wise gated layer and a residual binding interaction convolution layer. This allows the model to control the information flow in peptides to be aware of anchor positions and model the interactions between peptides and the MHC pseudo (binding) sequence directly with multiple convolutional kernels.</p><p><strong>Results: </strong>The performance of DeepMHCI has been thoroughly validated by extensive experiments on four benchmark datasets under various settings, such as 5-fold cross-validation, validation with the independent testing set, external HPV vaccine identification, and external CD8+ epitope identification. Experimental results with visualization of binding motifs demonstrate that DeepMHCI outperformed all competing methods, especially on non-9-mer peptides binding prediction.</p><p><strong>Availability and implementation: </strong>DeepMHCI is publicly available at https://github.com/ZhuLab-Fudan/DeepMHCI.</p>","PeriodicalId":8903,"journal":{"name":"Bioinformatics","volume":" ","pages":""},"PeriodicalIF":5.4000,"publicationDate":"2023-09-02","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10516514/pdf/","citationCount":"0","resultStr":"{\"title\":\"DeepMHCI: an anchor position-aware deep interaction model for accurate MHC-I peptide binding affinity prediction.\",\"authors\":\"Wei Qu, Ronghui You, Hiroshi Mamitsuka, Shanfeng Zhu\",\"doi\":\"10.1093/bioinformatics/btad551\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Motivation: </strong>Computationally predicting major histocompatibility complex class I (MHC-I) peptide binding affinity is an important problem in immunological bioinformatics, which is also crucial for the identification of neoantigens for personalized therapeutic cancer vaccines. Recent cutting-edge deep learning-based methods for this problem cannot achieve satisfactory performance, especially for non-9-mer peptides. This is because such methods generate the input by simply concatenating the two given sequences: a peptide and (the pseudo sequence of) an MHC class I molecule, which cannot precisely capture the anchor positions of the MHC binding motif for the peptides with variable lengths. We thus developed an anchor position-aware and high-performance deep model, DeepMHCI, with a position-wise gated layer and a residual binding interaction convolution layer. This allows the model to control the information flow in peptides to be aware of anchor positions and model the interactions between peptides and the MHC pseudo (binding) sequence directly with multiple convolutional kernels.</p><p><strong>Results: </strong>The performance of DeepMHCI has been thoroughly validated by extensive experiments on four benchmark datasets under various settings, such as 5-fold cross-validation, validation with the independent testing set, external HPV vaccine identification, and external CD8+ epitope identification. Experimental results with visualization of binding motifs demonstrate that DeepMHCI outperformed all competing methods, especially on non-9-mer peptides binding prediction.</p><p><strong>Availability and implementation: </strong>DeepMHCI is publicly available at https://github.com/ZhuLab-Fudan/DeepMHCI.</p>\",\"PeriodicalId\":8903,\"journal\":{\"name\":\"Bioinformatics\",\"volume\":\" \",\"pages\":\"\"},\"PeriodicalIF\":5.4000,\"publicationDate\":\"2023-09-02\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10516514/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Bioinformatics\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://doi.org/10.1093/bioinformatics/btad551\",\"RegionNum\":3,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"BIOCHEMICAL RESEARCH METHODS\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Bioinformatics","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1093/bioinformatics/btad551","RegionNum":3,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"BIOCHEMICAL RESEARCH METHODS","Score":null,"Total":0}
DeepMHCI: an anchor position-aware deep interaction model for accurate MHC-I peptide binding affinity prediction.
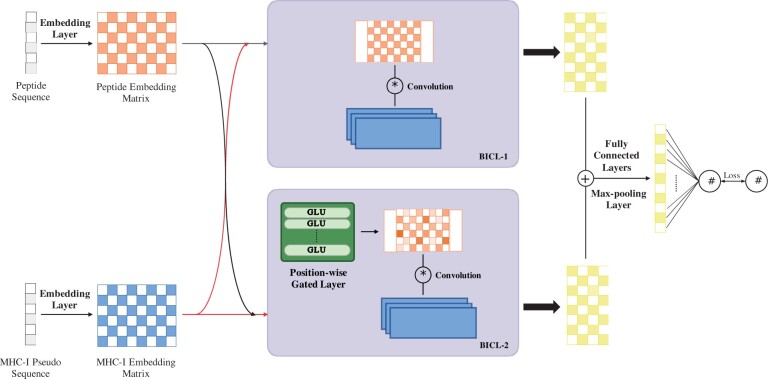
Motivation: Computationally predicting major histocompatibility complex class I (MHC-I) peptide binding affinity is an important problem in immunological bioinformatics, which is also crucial for the identification of neoantigens for personalized therapeutic cancer vaccines. Recent cutting-edge deep learning-based methods for this problem cannot achieve satisfactory performance, especially for non-9-mer peptides. This is because such methods generate the input by simply concatenating the two given sequences: a peptide and (the pseudo sequence of) an MHC class I molecule, which cannot precisely capture the anchor positions of the MHC binding motif for the peptides with variable lengths. We thus developed an anchor position-aware and high-performance deep model, DeepMHCI, with a position-wise gated layer and a residual binding interaction convolution layer. This allows the model to control the information flow in peptides to be aware of anchor positions and model the interactions between peptides and the MHC pseudo (binding) sequence directly with multiple convolutional kernels.
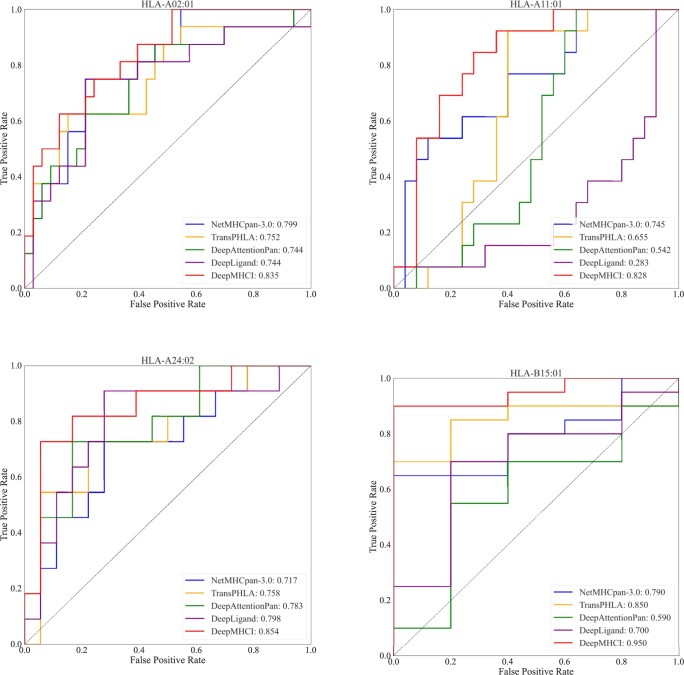
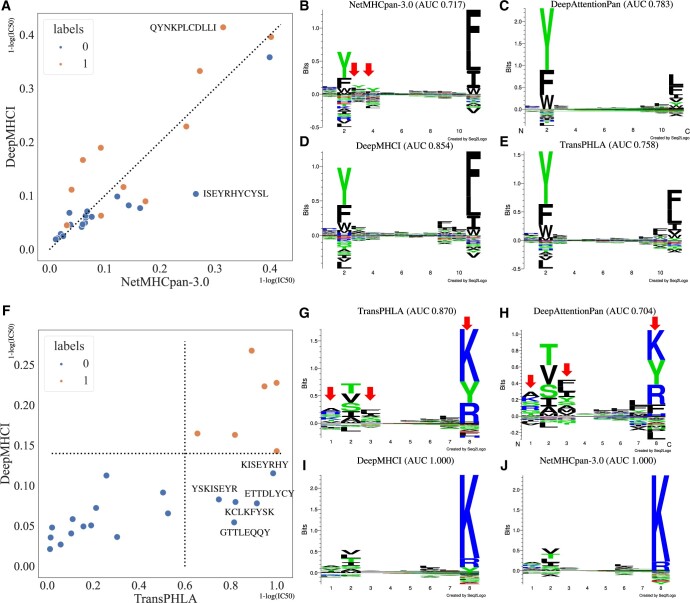
Results: The performance of DeepMHCI has been thoroughly validated by extensive experiments on four benchmark datasets under various settings, such as 5-fold cross-validation, validation with the independent testing set, external HPV vaccine identification, and external CD8+ epitope identification. Experimental results with visualization of binding motifs demonstrate that DeepMHCI outperformed all competing methods, especially on non-9-mer peptides binding prediction.
Availability and implementation: DeepMHCI is publicly available at https://github.com/ZhuLab-Fudan/DeepMHCI.
期刊介绍:
The leading journal in its field, Bioinformatics publishes the highest quality scientific papers and review articles of interest to academic and industrial researchers. Its main focus is on new developments in genome bioinformatics and computational biology. Two distinct sections within the journal - Discovery Notes and Application Notes- focus on shorter papers; the former reporting biologically interesting discoveries using computational methods, the latter exploring the applications used for experiments.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
