Brian Johnson, Yubo Shuai, Jason Schweinsberg, Kit Curtius
{"title":"cloneRate:使用联合理论快速估计单细胞克隆动力学。","authors":"Brian Johnson, Yubo Shuai, Jason Schweinsberg, Kit Curtius","doi":"10.1093/bioinformatics/btad561","DOIUrl":null,"url":null,"abstract":"<p><strong>Motivation: </strong>While evolutionary approaches to medicine show promise, measuring evolution itself is difficult due to experimental constraints and the dynamic nature of body systems. In cancer evolution, continuous observation of clonal architecture is impossible, and longitudinal samples from multiple timepoints are rare. Increasingly available DNA sequencing datasets at single-cell resolution enable the reconstruction of past evolution using mutational history, allowing for a better understanding of dynamics prior to detectable disease. There is an unmet need for an accurate, fast, and easy-to-use method to quantify clone growth dynamics from these datasets.</p><p><strong>Results: </strong>We derived methods based on coalescent theory for estimating the net growth rate of clones using either reconstructed phylogenies or the number of shared mutations. We applied and validated our analytical methods for estimating the net growth rate of clones, eliminating the need for complex simulations used in previous methods. When applied to hematopoietic data, we show that our estimates may have broad applications to improve mechanistic understanding and prognostic ability. Compared to clones with a single or unknown driver mutation, clones with multiple drivers have significantly increased growth rates (median 0.94 versus 0.25 per year; P = 1.6×10-6). Further, stratifying patients with a myeloproliferative neoplasm (MPN) by the growth rate of their fittest clone shows that higher growth rates are associated with shorter time to MPN diagnosis (median 13.9 versus 26.4 months; P = 0.0026).</p><p><strong>Availability and implementation: </strong>We developed a publicly available R package, cloneRate, to implement our methods (Package website: https://bdj34.github.io/cloneRate/). Source code: https://github.com/bdj34/cloneRate/.</p>","PeriodicalId":8903,"journal":{"name":"Bioinformatics","volume":" ","pages":""},"PeriodicalIF":5.4000,"publicationDate":"2023-09-02","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10534056/pdf/","citationCount":"0","resultStr":"{\"title\":\"cloneRate: fast estimation of single-cell clonal dynamics using coalescent theory.\",\"authors\":\"Brian Johnson, Yubo Shuai, Jason Schweinsberg, Kit Curtius\",\"doi\":\"10.1093/bioinformatics/btad561\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Motivation: </strong>While evolutionary approaches to medicine show promise, measuring evolution itself is difficult due to experimental constraints and the dynamic nature of body systems. In cancer evolution, continuous observation of clonal architecture is impossible, and longitudinal samples from multiple timepoints are rare. Increasingly available DNA sequencing datasets at single-cell resolution enable the reconstruction of past evolution using mutational history, allowing for a better understanding of dynamics prior to detectable disease. There is an unmet need for an accurate, fast, and easy-to-use method to quantify clone growth dynamics from these datasets.</p><p><strong>Results: </strong>We derived methods based on coalescent theory for estimating the net growth rate of clones using either reconstructed phylogenies or the number of shared mutations. We applied and validated our analytical methods for estimating the net growth rate of clones, eliminating the need for complex simulations used in previous methods. When applied to hematopoietic data, we show that our estimates may have broad applications to improve mechanistic understanding and prognostic ability. Compared to clones with a single or unknown driver mutation, clones with multiple drivers have significantly increased growth rates (median 0.94 versus 0.25 per year; P = 1.6×10-6). Further, stratifying patients with a myeloproliferative neoplasm (MPN) by the growth rate of their fittest clone shows that higher growth rates are associated with shorter time to MPN diagnosis (median 13.9 versus 26.4 months; P = 0.0026).</p><p><strong>Availability and implementation: </strong>We developed a publicly available R package, cloneRate, to implement our methods (Package website: https://bdj34.github.io/cloneRate/). Source code: https://github.com/bdj34/cloneRate/.</p>\",\"PeriodicalId\":8903,\"journal\":{\"name\":\"Bioinformatics\",\"volume\":\" \",\"pages\":\"\"},\"PeriodicalIF\":5.4000,\"publicationDate\":\"2023-09-02\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10534056/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Bioinformatics\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://doi.org/10.1093/bioinformatics/btad561\",\"RegionNum\":3,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"BIOCHEMICAL RESEARCH METHODS\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Bioinformatics","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1093/bioinformatics/btad561","RegionNum":3,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"BIOCHEMICAL RESEARCH METHODS","Score":null,"Total":0}
cloneRate: fast estimation of single-cell clonal dynamics using coalescent theory.
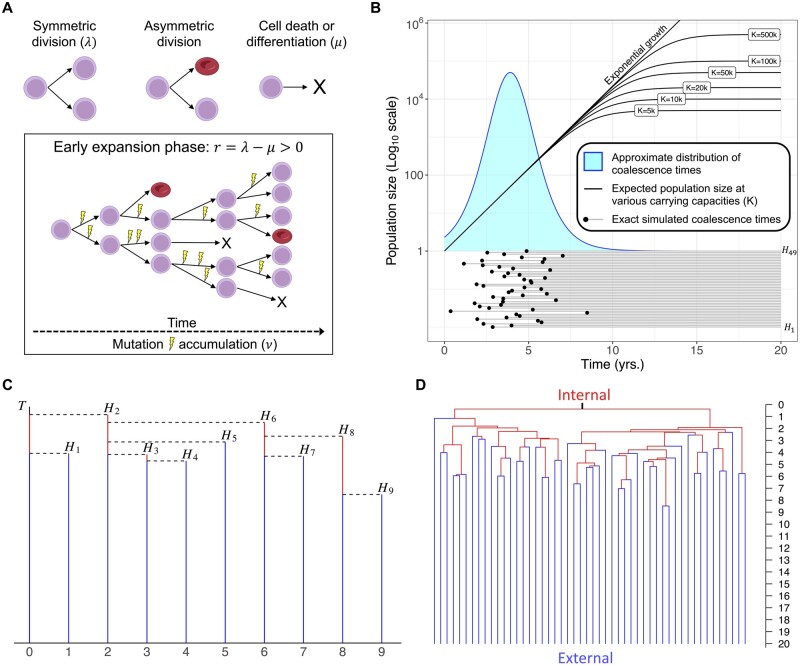
Motivation: While evolutionary approaches to medicine show promise, measuring evolution itself is difficult due to experimental constraints and the dynamic nature of body systems. In cancer evolution, continuous observation of clonal architecture is impossible, and longitudinal samples from multiple timepoints are rare. Increasingly available DNA sequencing datasets at single-cell resolution enable the reconstruction of past evolution using mutational history, allowing for a better understanding of dynamics prior to detectable disease. There is an unmet need for an accurate, fast, and easy-to-use method to quantify clone growth dynamics from these datasets.
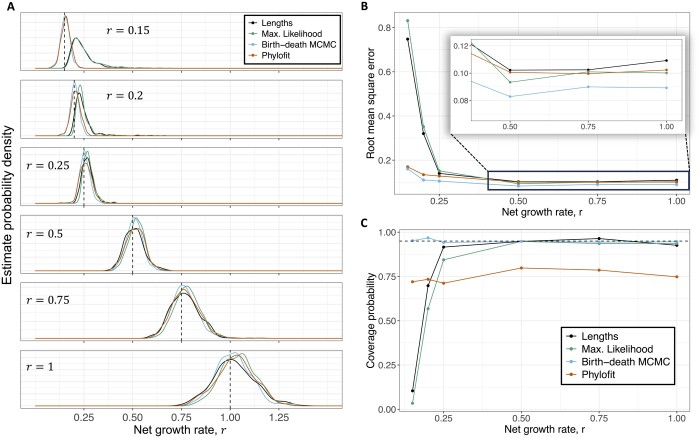
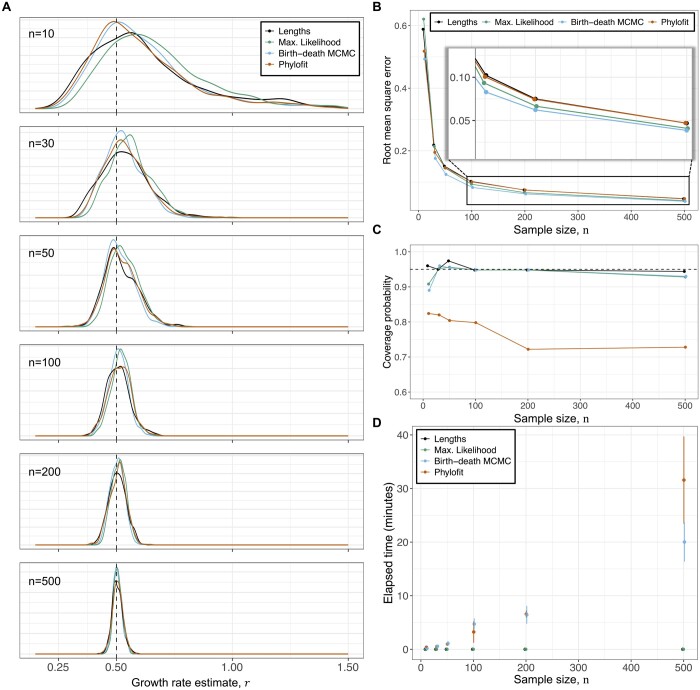
Results: We derived methods based on coalescent theory for estimating the net growth rate of clones using either reconstructed phylogenies or the number of shared mutations. We applied and validated our analytical methods for estimating the net growth rate of clones, eliminating the need for complex simulations used in previous methods. When applied to hematopoietic data, we show that our estimates may have broad applications to improve mechanistic understanding and prognostic ability. Compared to clones with a single or unknown driver mutation, clones with multiple drivers have significantly increased growth rates (median 0.94 versus 0.25 per year; P = 1.6×10-6). Further, stratifying patients with a myeloproliferative neoplasm (MPN) by the growth rate of their fittest clone shows that higher growth rates are associated with shorter time to MPN diagnosis (median 13.9 versus 26.4 months; P = 0.0026).
Availability and implementation: We developed a publicly available R package, cloneRate, to implement our methods (Package website: https://bdj34.github.io/cloneRate/). Source code: https://github.com/bdj34/cloneRate/.
期刊介绍:
The leading journal in its field, Bioinformatics publishes the highest quality scientific papers and review articles of interest to academic and industrial researchers. Its main focus is on new developments in genome bioinformatics and computational biology. Two distinct sections within the journal - Discovery Notes and Application Notes- focus on shorter papers; the former reporting biologically interesting discoveries using computational methods, the latter exploring the applications used for experiments.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
