{"title":"用环磷酰胺治疗严重隐性萎缩性表皮松解症儿童的肾病:病例报告。","authors":"Cahyani Gita Ambarsari, Retno Palupi-Baroto, Fira Alyssa Gabriella Sinuraya, Elvi Suryati, Etty Widyastuti, Suci Widhiati","doi":"10.1159/000530875","DOIUrl":null,"url":null,"abstract":"<p><p>Long-term inflammation and recurrent skin infection in recessive dystrophic epidermolysis bullosa (RDEB) are associated with the presence of immunoglobulin A (IgA)-containing immune complexes in the glomerulus. Only eight pediatric RDEB cases with IgA nephropathy (IgAN) have been documented in English-language literature. Most RDEB patients with IgAN progress to kidney failure within 5 years of diagnosis, indicating that these patients may require more intensive early treatment compared to those with primary IgAN. However, diagnosing IgAN in RDEB cases with severe cutaneous manifestations can be challenging. Herein, we report a rare case of nephropathy in an 11-year-old boy with severe RDEB and a frameshift mutation on the <i>COL7A1</i> gene, which may manifest as kidney disorders. He presented with persistent hematuria and progressing proteinuria. A presumptive IgAN diagnosis was based on clinical features and increased IgA serum levels, as kidney biopsy was refused by his parents. Nephrotic-range proteinuria persisted despite initial steroid and lisinopril treatment. Monthly intravenous cyclophosphamide (IV CPA; 500 mg/m<sup>2</sup>) led to proteinuria remission and preservation of kidney function for 2 years posttreatment. We conclude that <i>COL7A1</i> mutations may result in extracutaneous manifestations, including kidney disorders. The association between IgA-containing immune complex deposits in the glomerulus and recurrent skin infection in RDEB may indicate IgAN, particularly when kidney biopsy is infeasible due to severe skin manifestations. In our case, positive results with IV CPA suggest further investigation is needed to explore its potential role in non-rapidly progressing IgAN in children with RDEB.</p>","PeriodicalId":9599,"journal":{"name":"Case Reports in Nephrology and Dialysis","volume":"13 1","pages":"75-83"},"PeriodicalIF":0.9000,"publicationDate":"2023-07-14","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/94/a2/cnd-2023-0013-0001-530875.PMC10359707.pdf","citationCount":"0","resultStr":"{\"title\":\"Nephropathy in a Child with Severe Recessive Dystrophic Epidermolysis Bullosa Treated with Cyclophosphamide: A Case Report.\",\"authors\":\"Cahyani Gita Ambarsari, Retno Palupi-Baroto, Fira Alyssa Gabriella Sinuraya, Elvi Suryati, Etty Widyastuti, Suci Widhiati\",\"doi\":\"10.1159/000530875\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Long-term inflammation and recurrent skin infection in recessive dystrophic epidermolysis bullosa (RDEB) are associated with the presence of immunoglobulin A (IgA)-containing immune complexes in the glomerulus. Only eight pediatric RDEB cases with IgA nephropathy (IgAN) have been documented in English-language literature. Most RDEB patients with IgAN progress to kidney failure within 5 years of diagnosis, indicating that these patients may require more intensive early treatment compared to those with primary IgAN. However, diagnosing IgAN in RDEB cases with severe cutaneous manifestations can be challenging. Herein, we report a rare case of nephropathy in an 11-year-old boy with severe RDEB and a frameshift mutation on the <i>COL7A1</i> gene, which may manifest as kidney disorders. He presented with persistent hematuria and progressing proteinuria. A presumptive IgAN diagnosis was based on clinical features and increased IgA serum levels, as kidney biopsy was refused by his parents. Nephrotic-range proteinuria persisted despite initial steroid and lisinopril treatment. Monthly intravenous cyclophosphamide (IV CPA; 500 mg/m<sup>2</sup>) led to proteinuria remission and preservation of kidney function for 2 years posttreatment. We conclude that <i>COL7A1</i> mutations may result in extracutaneous manifestations, including kidney disorders. The association between IgA-containing immune complex deposits in the glomerulus and recurrent skin infection in RDEB may indicate IgAN, particularly when kidney biopsy is infeasible due to severe skin manifestations. In our case, positive results with IV CPA suggest further investigation is needed to explore its potential role in non-rapidly progressing IgAN in children with RDEB.</p>\",\"PeriodicalId\":9599,\"journal\":{\"name\":\"Case Reports in Nephrology and Dialysis\",\"volume\":\"13 1\",\"pages\":\"75-83\"},\"PeriodicalIF\":0.9000,\"publicationDate\":\"2023-07-14\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/94/a2/cnd-2023-0013-0001-530875.PMC10359707.pdf\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Case Reports in Nephrology and Dialysis\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1159/000530875\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2023/1/1 0:00:00\",\"PubModel\":\"eCollection\",\"JCR\":\"Q4\",\"JCRName\":\"UROLOGY & NEPHROLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Case Reports in Nephrology and Dialysis","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1159/000530875","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2023/1/1 0:00:00","PubModel":"eCollection","JCR":"Q4","JCRName":"UROLOGY & NEPHROLOGY","Score":null,"Total":0}
Nephropathy in a Child with Severe Recessive Dystrophic Epidermolysis Bullosa Treated with Cyclophosphamide: A Case Report.
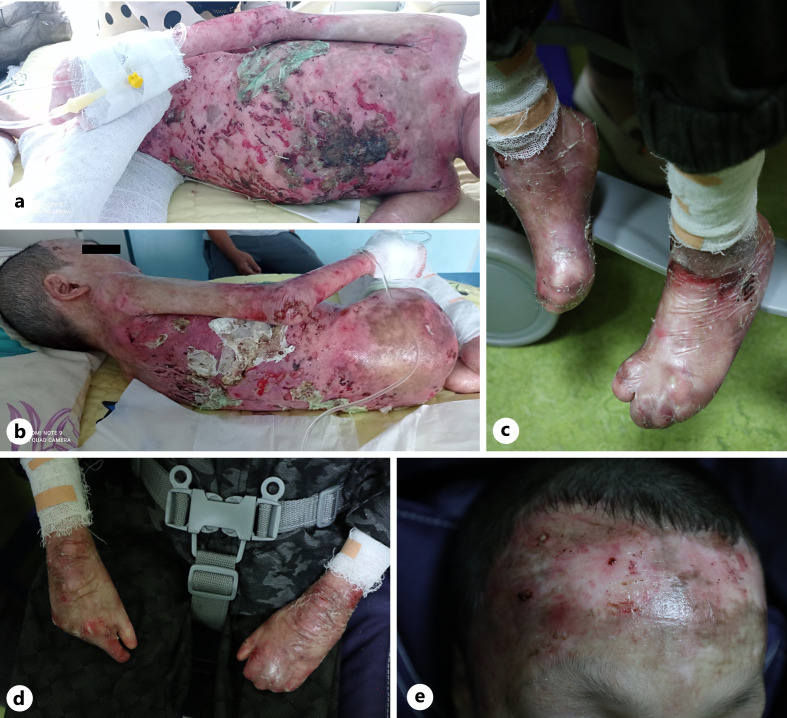
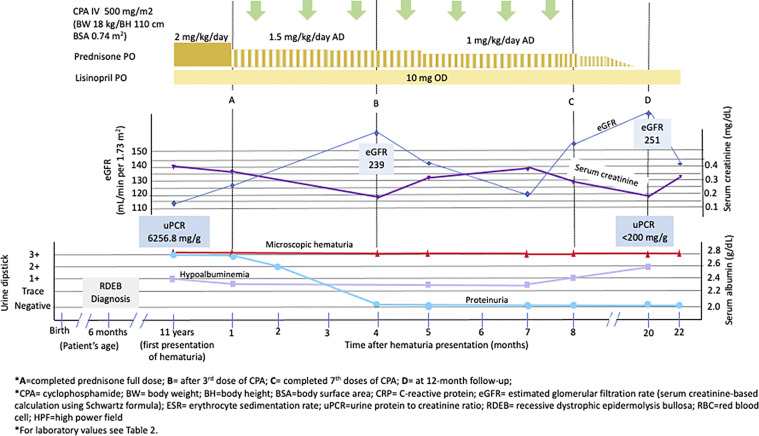
Long-term inflammation and recurrent skin infection in recessive dystrophic epidermolysis bullosa (RDEB) are associated with the presence of immunoglobulin A (IgA)-containing immune complexes in the glomerulus. Only eight pediatric RDEB cases with IgA nephropathy (IgAN) have been documented in English-language literature. Most RDEB patients with IgAN progress to kidney failure within 5 years of diagnosis, indicating that these patients may require more intensive early treatment compared to those with primary IgAN. However, diagnosing IgAN in RDEB cases with severe cutaneous manifestations can be challenging. Herein, we report a rare case of nephropathy in an 11-year-old boy with severe RDEB and a frameshift mutation on the COL7A1 gene, which may manifest as kidney disorders. He presented with persistent hematuria and progressing proteinuria. A presumptive IgAN diagnosis was based on clinical features and increased IgA serum levels, as kidney biopsy was refused by his parents. Nephrotic-range proteinuria persisted despite initial steroid and lisinopril treatment. Monthly intravenous cyclophosphamide (IV CPA; 500 mg/m2) led to proteinuria remission and preservation of kidney function for 2 years posttreatment. We conclude that COL7A1 mutations may result in extracutaneous manifestations, including kidney disorders. The association between IgA-containing immune complex deposits in the glomerulus and recurrent skin infection in RDEB may indicate IgAN, particularly when kidney biopsy is infeasible due to severe skin manifestations. In our case, positive results with IV CPA suggest further investigation is needed to explore its potential role in non-rapidly progressing IgAN in children with RDEB.
期刊介绍:
This peer-reviewed online-only journal publishes original case reports covering the entire spectrum of nephrology and dialysis, including genetic susceptibility, clinical presentation, diagnosis, treatment or prevention, toxicities of therapy, critical care, supportive care, quality-of-life and survival issues. The journal will also accept case reports dealing with the use of novel technologies, both in the arena of diagnosis and treatment. Supplementary material is welcomed.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
