Nana Li, Hong Kang, Yanna Zou, Zhen Liu, Ying Deng, Meixian Wang, Lu Li, Hong Qin, Xiaoqiong Qiu, Yanping Wang, Jun Zhu, Mark Agostino, Julian I-T Heng, Ping Yu
{"title":"一个非综合征性智力残疾家族中一个新的杂合子ZBTB18错义突变。","authors":"Nana Li, Hong Kang, Yanna Zou, Zhen Liu, Ying Deng, Meixian Wang, Lu Li, Hong Qin, Xiaoqiong Qiu, Yanping Wang, Jun Zhu, Mark Agostino, Julian I-T Heng, Ping Yu","doi":"10.1007/s10048-023-00727-7","DOIUrl":null,"url":null,"abstract":"<p><p>Intellectual disability (ID) is a common neurodevelopmental disorder characterized by significantly impaired adaptive behavior and cognitive capacity. High throughput sequencing approaches have revealed the genetic etiologies for 25-50% of ID patients, while inherited genetic mutations were detected in <5% cases. Here, we investigated the genetic cause for non-syndromic ID in a Han Chinese family. Whole genome sequencing was performed on identical twin sisters diagnosed with ID, their respective children, and their asymptomatic parents. Data was filtered for rare variants, and in silico prediction tools were used to establish pathogenic alleles. Candidate mutations were validated by Sanger sequencing. In silico modeling was used to evaluate the mutation's effects on the protein encoded by a candidate coding gene. A novel heterozygous variant in the ZBTB18 gene c.1323C>G (p.His441Gln) was identified. This variant co-segregated with affected individuals in an autosomal dominant pattern and was not detected in asymptomatic family members. Molecular studies reveal that a p.His441Gln substitution disrupts zinc binding within the second zinc finger and disrupts the capacity for ZBTB18 to bind DNA. This is the first report of an inherited ZBTB18 mutation for ID. This study further validates WGS for the accurate molecular diagnosis of ID.</p>","PeriodicalId":56106,"journal":{"name":"Neurogenetics","volume":" ","pages":"251-262"},"PeriodicalIF":1.2000,"publicationDate":"2023-10-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"A novel heterozygous ZBTB18 missense mutation in a family with non-syndromic intellectual disability.\",\"authors\":\"Nana Li, Hong Kang, Yanna Zou, Zhen Liu, Ying Deng, Meixian Wang, Lu Li, Hong Qin, Xiaoqiong Qiu, Yanping Wang, Jun Zhu, Mark Agostino, Julian I-T Heng, Ping Yu\",\"doi\":\"10.1007/s10048-023-00727-7\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Intellectual disability (ID) is a common neurodevelopmental disorder characterized by significantly impaired adaptive behavior and cognitive capacity. High throughput sequencing approaches have revealed the genetic etiologies for 25-50% of ID patients, while inherited genetic mutations were detected in <5% cases. Here, we investigated the genetic cause for non-syndromic ID in a Han Chinese family. Whole genome sequencing was performed on identical twin sisters diagnosed with ID, their respective children, and their asymptomatic parents. Data was filtered for rare variants, and in silico prediction tools were used to establish pathogenic alleles. Candidate mutations were validated by Sanger sequencing. In silico modeling was used to evaluate the mutation's effects on the protein encoded by a candidate coding gene. A novel heterozygous variant in the ZBTB18 gene c.1323C>G (p.His441Gln) was identified. This variant co-segregated with affected individuals in an autosomal dominant pattern and was not detected in asymptomatic family members. Molecular studies reveal that a p.His441Gln substitution disrupts zinc binding within the second zinc finger and disrupts the capacity for ZBTB18 to bind DNA. This is the first report of an inherited ZBTB18 mutation for ID. This study further validates WGS for the accurate molecular diagnosis of ID.</p>\",\"PeriodicalId\":56106,\"journal\":{\"name\":\"Neurogenetics\",\"volume\":\" \",\"pages\":\"251-262\"},\"PeriodicalIF\":1.2000,\"publicationDate\":\"2023-10-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Neurogenetics\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://doi.org/10.1007/s10048-023-00727-7\",\"RegionNum\":4,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2023/7/31 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"Q3\",\"JCRName\":\"CLINICAL NEUROLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Neurogenetics","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1007/s10048-023-00727-7","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2023/7/31 0:00:00","PubModel":"Epub","JCR":"Q3","JCRName":"CLINICAL NEUROLOGY","Score":null,"Total":0}
A novel heterozygous ZBTB18 missense mutation in a family with non-syndromic intellectual disability.
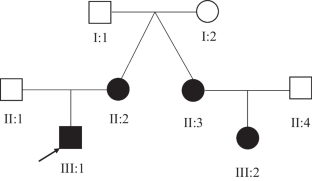
Intellectual disability (ID) is a common neurodevelopmental disorder characterized by significantly impaired adaptive behavior and cognitive capacity. High throughput sequencing approaches have revealed the genetic etiologies for 25-50% of ID patients, while inherited genetic mutations were detected in <5% cases. Here, we investigated the genetic cause for non-syndromic ID in a Han Chinese family. Whole genome sequencing was performed on identical twin sisters diagnosed with ID, their respective children, and their asymptomatic parents. Data was filtered for rare variants, and in silico prediction tools were used to establish pathogenic alleles. Candidate mutations were validated by Sanger sequencing. In silico modeling was used to evaluate the mutation's effects on the protein encoded by a candidate coding gene. A novel heterozygous variant in the ZBTB18 gene c.1323C>G (p.His441Gln) was identified. This variant co-segregated with affected individuals in an autosomal dominant pattern and was not detected in asymptomatic family members. Molecular studies reveal that a p.His441Gln substitution disrupts zinc binding within the second zinc finger and disrupts the capacity for ZBTB18 to bind DNA. This is the first report of an inherited ZBTB18 mutation for ID. This study further validates WGS for the accurate molecular diagnosis of ID.
期刊介绍:
Neurogenetics publishes findings that contribute to a better understanding of the genetic basis of normal and abnormal function of the nervous system. Neurogenetic disorders are the main focus of the journal. Neurogenetics therefore includes findings in humans and other organisms that help understand neurological disease mechanisms and publishes papers from many different fields such as biophysics, cell biology, human genetics, neuroanatomy, neurochemistry, neurology, neuropathology, neurosurgery and psychiatry.
All papers submitted to Neurogenetics should be of sufficient immediate importance to justify urgent publication. They should present new scientific results. Data merely confirming previously published findings are not acceptable.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
