{"title":"E148Q突变继发于家族性地中海热的淀粉样甲状腺肿:一个独特的病例","authors":"Juan C. A. Moreno, Eduardo Eyzaguirre, Suimin Qiu","doi":"10.1002/cdt3.79","DOIUrl":null,"url":null,"abstract":"<p>Dear Editor,</p><p>Goiter is defined as the enlargement of the thyroid gland. It is currently divided into diffuse and nodular and subdivided into toxic (associated with hyperthyroidism) or nontoxic (associated with normal thyroid stimulating hormone [TSH] levels).<span><sup>1</sup></span> The most common cause of goiter is iodine deficiency,<span><sup>2</sup></span> and other causes are increased levels of TSH, natural goitrogens, iron and vitamin A deficiency, genetic factors (<i>DICER1</i> syndrome and <i>PTEN</i> hamartoma tumor syndrome), and hereditary (Plummer syndrome) factors.<span><sup>3</sup></span></p><p>A rare entity known as familial Mediterranean fever (FMF) can present with amyloid goiter. This is an autoimmune disorder that affects the Mediterranean littoral.<span><sup>4</sup></span> These patients present with fevers and abdominal and chest pain. This condition can produce fibrillar depositions of amyloid protein, usually affecting the kidney<span><sup>5</sup></span> but rarely involving the thyroid. We present a case of a 21-year-old woman with no medical history who presented to our hospital with a nontoxic diffuse goiter with initial presentation and pathologically confirmed as amyloid deposition secondary to FMF.</p><p>The patient is a 21-year-old Asian American woman with no significant medical history. She presented to our institution with dyspnea and dysphagia. An ultrasound from an outside hospital revealed diffuse thyroid enlargement. Our in-house laboratory results showed normal TSH, T4, T3, and elevated TPO antibody and erythrocyte sedimentation rate. A CT scan was performed on the patient showing heterogenous thyromegaly wrapping around the trachea and esophagus (Figure 1). A fine needle aspirate was performed, which showed a fibroinflammatory lesion. The patient started on steroids with no improvement, and a total thyroidectomy was performed. Macroscopic examination revealed a poorly defined, firm mass with pale tan areas of discoloration (Figure 2). Microscopic examination revealed an atrophic thyroid parenchyma with diffuse adipose cell metaplasia and diffuse interfollicular deposition of acellular amorphous material consistent with amyloid (Figure 3). Multifocal areas of chronic inflammation were also seen. Congo red stain was positive for amyloid (Figure 4A) with apple-green birefringence under polarized light (Figure 4B). Liquid chromatography tandem mass spectrometry was performed on peptides extracted from the Congo red-positive/microdissected areas of the paraffin-embedded thyroid specimen. The detected peptide profile was consistent with AA (SAA)-type amyloid deposition. The patient consequently had genetic testing showing a homozygous E148Q mutation. This confirms the clinical syndrome of FMF. After continuous follow-up, 10 years later, she developed end-stage renal failure with a renal biopsy confirming Renal AA amyloidosis. Since then, she has been on hemodialysis and is now on the waiting list for a renal transplant.</p><p>Amyloid goiter is only seen in 0.04% of patients with systemic amyloidosis.<span><sup>6</sup></span> Amyloid depositions may be localized or systemic, as in our patient who developed both but initially presented as localized goiter. The International Society of Amyloidosis guidelines defines amyloid as an extracellular deposition of a fibrillary protein that is recognizable by its affinity for Congo red and its apple-green birefringence under polarized light.<span><sup>7</sup></span> We confirm the amyloid depositions in our case with the latter ancillary studies.</p><p>To date, there are 36 proteins identified as amyloidogenic in humans. AL, which is amyloidosis derived from immunoglobulin light chain, is the most common presentation of systemic amyloidosis in the developed world.<span><sup>8</sup></span> In our case, we identified a cause of AA-type amyloid deposition, amyloid fibrils derived from serum amyloid A protein, which is the leading cause of systemic amyloidosis in developing countries.<span><sup>9</sup></span> The AA protein is associated with hereditary autoimmune diseases, such as, in our case, FMF.</p><p>FMF has autosomal recessive inheritance with gene polymorphism and can result in amyloidosis. This entity results from the gain of function mutations of the Mediterranean fever gene (<i>MEFV</i>) located on chromosome 16 (16p13.3).<span><sup>10</sup></span> MEFV gene has 10 exons, and there are more than 370 variants identified to date. The most pathogenic mutations in FMF are M694V, M694I, M680I, and V726A in exon 10,<span><sup>11</sup></span> which were not found in our case. The E148Q variant in exon 2 is one of the most common substitutions in the <i>MEFV</i> gene<span><sup>11</sup></span> in most FMF cases. It occurs as the only identified variant or in parallel with other variants, including exon 10 mutations. Limited evidence supports that homozygous E148Q is a disease-causing mutation. In our patient, this was the variant detected. The E148Q variant in exon 2 is found frequently in Japanese patients with FMF,<span><sup>12</sup></span> which possibly correlates with our patient's Asian American heritage. According to the literature, patients with homozygous E148Q variant may present with late-onset and mild disease,<span><sup>13, 14</sup></span> although none of these cases presented with amyloid goiter. The exact prevalence of amyloid goiter in this entity is yet to be established. Approximately 15.00% was found by Vergneault et al.,<span><sup>15</sup></span> 45.00% in the Ozdemir study,<span><sup>16</sup></span> and 0.27% in Altiparmak et al.<span><sup>17</sup></span> findings. None of the previous reports had genetic testing on the patients. Most of these cases had other systemic symptoms, renal failure being the most common. Our patient developed renal failure in a prospective fashion.</p><p>This case of isolated amyloid goiter is a unique presentation of FMF. Despite the literature having reports of amyloid goiter, most of the patients had other systemic diseases at the time of presentation of the disease. The homozygous E148Q variant in this case is interesting, due to its mild disease presentation and lack of association with amyloid goiter in the literature. It is important to recognize that diffuse goiter can be the early clinical presentation of FMF.</p><p>Juan C. A. Moreno worked on the case report conception and contributed to the data collection. Juan C. A. Moreno, Suimin Qiu, and Eduardo Eyzaguirre contributed to the pathological slides review and data analysis. Juan C. A. Moreno was responsible for getting the CT images from the medical records of the hospital. Eduardo Eyzaguirre and Suimin Qiu provided explanations about the case reported. Juan C. A. Moreno worked on the histology figures, figure illustrations, and case study timeline presentation. Suimin Qiu was responsible for the study supervision. All authors critically revised and edited the manuscript before approving the final draft of the manuscript.</p><p>The authors declare no conflict of interest.</p><p>The work has been carried out in accordance with the code of ethics of the world medical association (<i>Declaration of Helsinki</i>).</p>","PeriodicalId":32096,"journal":{"name":"Chronic Diseases and Translational Medicine","volume":"9 3","pages":"266-268"},"PeriodicalIF":0.0000,"publicationDate":"2023-06-14","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/37/d6/CDT3-9-266.PMC10497809.pdf","citationCount":"0","resultStr":"{\"title\":\"Amyloid goiter secondary to familial Mediterranean fever with E148Q mutation: A unique case\",\"authors\":\"Juan C. A. Moreno, Eduardo Eyzaguirre, Suimin Qiu\",\"doi\":\"10.1002/cdt3.79\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p>Dear Editor,</p><p>Goiter is defined as the enlargement of the thyroid gland. It is currently divided into diffuse and nodular and subdivided into toxic (associated with hyperthyroidism) or nontoxic (associated with normal thyroid stimulating hormone [TSH] levels).<span><sup>1</sup></span> The most common cause of goiter is iodine deficiency,<span><sup>2</sup></span> and other causes are increased levels of TSH, natural goitrogens, iron and vitamin A deficiency, genetic factors (<i>DICER1</i> syndrome and <i>PTEN</i> hamartoma tumor syndrome), and hereditary (Plummer syndrome) factors.<span><sup>3</sup></span></p><p>A rare entity known as familial Mediterranean fever (FMF) can present with amyloid goiter. This is an autoimmune disorder that affects the Mediterranean littoral.<span><sup>4</sup></span> These patients present with fevers and abdominal and chest pain. This condition can produce fibrillar depositions of amyloid protein, usually affecting the kidney<span><sup>5</sup></span> but rarely involving the thyroid. We present a case of a 21-year-old woman with no medical history who presented to our hospital with a nontoxic diffuse goiter with initial presentation and pathologically confirmed as amyloid deposition secondary to FMF.</p><p>The patient is a 21-year-old Asian American woman with no significant medical history. She presented to our institution with dyspnea and dysphagia. An ultrasound from an outside hospital revealed diffuse thyroid enlargement. Our in-house laboratory results showed normal TSH, T4, T3, and elevated TPO antibody and erythrocyte sedimentation rate. A CT scan was performed on the patient showing heterogenous thyromegaly wrapping around the trachea and esophagus (Figure 1). A fine needle aspirate was performed, which showed a fibroinflammatory lesion. The patient started on steroids with no improvement, and a total thyroidectomy was performed. Macroscopic examination revealed a poorly defined, firm mass with pale tan areas of discoloration (Figure 2). Microscopic examination revealed an atrophic thyroid parenchyma with diffuse adipose cell metaplasia and diffuse interfollicular deposition of acellular amorphous material consistent with amyloid (Figure 3). Multifocal areas of chronic inflammation were also seen. Congo red stain was positive for amyloid (Figure 4A) with apple-green birefringence under polarized light (Figure 4B). Liquid chromatography tandem mass spectrometry was performed on peptides extracted from the Congo red-positive/microdissected areas of the paraffin-embedded thyroid specimen. The detected peptide profile was consistent with AA (SAA)-type amyloid deposition. The patient consequently had genetic testing showing a homozygous E148Q mutation. This confirms the clinical syndrome of FMF. After continuous follow-up, 10 years later, she developed end-stage renal failure with a renal biopsy confirming Renal AA amyloidosis. Since then, she has been on hemodialysis and is now on the waiting list for a renal transplant.</p><p>Amyloid goiter is only seen in 0.04% of patients with systemic amyloidosis.<span><sup>6</sup></span> Amyloid depositions may be localized or systemic, as in our patient who developed both but initially presented as localized goiter. The International Society of Amyloidosis guidelines defines amyloid as an extracellular deposition of a fibrillary protein that is recognizable by its affinity for Congo red and its apple-green birefringence under polarized light.<span><sup>7</sup></span> We confirm the amyloid depositions in our case with the latter ancillary studies.</p><p>To date, there are 36 proteins identified as amyloidogenic in humans. AL, which is amyloidosis derived from immunoglobulin light chain, is the most common presentation of systemic amyloidosis in the developed world.<span><sup>8</sup></span> In our case, we identified a cause of AA-type amyloid deposition, amyloid fibrils derived from serum amyloid A protein, which is the leading cause of systemic amyloidosis in developing countries.<span><sup>9</sup></span> The AA protein is associated with hereditary autoimmune diseases, such as, in our case, FMF.</p><p>FMF has autosomal recessive inheritance with gene polymorphism and can result in amyloidosis. This entity results from the gain of function mutations of the Mediterranean fever gene (<i>MEFV</i>) located on chromosome 16 (16p13.3).<span><sup>10</sup></span> MEFV gene has 10 exons, and there are more than 370 variants identified to date. The most pathogenic mutations in FMF are M694V, M694I, M680I, and V726A in exon 10,<span><sup>11</sup></span> which were not found in our case. The E148Q variant in exon 2 is one of the most common substitutions in the <i>MEFV</i> gene<span><sup>11</sup></span> in most FMF cases. It occurs as the only identified variant or in parallel with other variants, including exon 10 mutations. Limited evidence supports that homozygous E148Q is a disease-causing mutation. In our patient, this was the variant detected. The E148Q variant in exon 2 is found frequently in Japanese patients with FMF,<span><sup>12</sup></span> which possibly correlates with our patient's Asian American heritage. According to the literature, patients with homozygous E148Q variant may present with late-onset and mild disease,<span><sup>13, 14</sup></span> although none of these cases presented with amyloid goiter. The exact prevalence of amyloid goiter in this entity is yet to be established. Approximately 15.00% was found by Vergneault et al.,<span><sup>15</sup></span> 45.00% in the Ozdemir study,<span><sup>16</sup></span> and 0.27% in Altiparmak et al.<span><sup>17</sup></span> findings. None of the previous reports had genetic testing on the patients. Most of these cases had other systemic symptoms, renal failure being the most common. Our patient developed renal failure in a prospective fashion.</p><p>This case of isolated amyloid goiter is a unique presentation of FMF. Despite the literature having reports of amyloid goiter, most of the patients had other systemic diseases at the time of presentation of the disease. The homozygous E148Q variant in this case is interesting, due to its mild disease presentation and lack of association with amyloid goiter in the literature. It is important to recognize that diffuse goiter can be the early clinical presentation of FMF.</p><p>Juan C. A. Moreno worked on the case report conception and contributed to the data collection. Juan C. A. Moreno, Suimin Qiu, and Eduardo Eyzaguirre contributed to the pathological slides review and data analysis. Juan C. A. Moreno was responsible for getting the CT images from the medical records of the hospital. Eduardo Eyzaguirre and Suimin Qiu provided explanations about the case reported. Juan C. A. Moreno worked on the histology figures, figure illustrations, and case study timeline presentation. Suimin Qiu was responsible for the study supervision. All authors critically revised and edited the manuscript before approving the final draft of the manuscript.</p><p>The authors declare no conflict of interest.</p><p>The work has been carried out in accordance with the code of ethics of the world medical association (<i>Declaration of Helsinki</i>).</p>\",\"PeriodicalId\":32096,\"journal\":{\"name\":\"Chronic Diseases and Translational Medicine\",\"volume\":\"9 3\",\"pages\":\"266-268\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2023-06-14\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/37/d6/CDT3-9-266.PMC10497809.pdf\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Chronic Diseases and Translational Medicine\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://onlinelibrary.wiley.com/doi/10.1002/cdt3.79\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"Medicine\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Chronic Diseases and Translational Medicine","FirstCategoryId":"3","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/cdt3.79","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"Medicine","Score":null,"Total":0}
引用次数: 0
摘要
亲爱的编辑,甲状腺肿的定义是甲状腺肿大。目前分为弥漫性和结节性,并细分为毒性(与甲状腺功能亢进有关)和无毒(与促甲状腺激素[TSH]水平正常有关)甲状腺肿最常见的原因是碘缺乏,其他原因包括TSH水平升高、天然甲状腺素、铁和维生素A缺乏、遗传因素(DICER1综合征和PTEN错构瘤肿瘤综合征)和遗传因素(Plummer综合征)。家族性地中海热(FMF)可表现为淀粉样甲状腺肿。这是一种影响地中海沿岸地区的自身免疫性疾病这些病人表现为发烧、腹痛和胸痛。此病可产生淀粉样蛋白的纤维状沉积,通常累及肾脏,但很少累及甲状腺。我们报告一例21岁无病史的女性,她以无毒弥漫性甲状腺肿来我院就诊,最初的表现和病理证实为继发于FMF的淀粉样蛋白沉积。患者为21岁亚裔美国女性,无明显病史。她以呼吸困难和吞咽困难来我们医院就诊。外院超声示弥漫性甲状腺肿大。我们的实验室结果显示TSH, T4, T3正常,TPO抗体和红细胞沉降率升高。患者行CT扫描,发现气管和食道周围有异质甲状腺肿大(图1)。行细针抽吸,发现纤维炎性病变。患者开始使用类固醇,但没有改善,并进行了甲状腺全切除术。宏观检查显示一个界限不清、坚硬的肿块,伴淡棕色变色区域(图2)。镜下检查显示萎缩性甲状腺实质伴弥漫性脂肪细胞化生,滤泡间弥漫性无细胞无定形物质沉积,与淀粉样蛋白一致(图3)。还可见多灶性慢性炎症。刚果红染色淀粉样蛋白阳性(图4A),偏振光下呈苹果绿双折射(图4B)。对从石蜡包埋甲状腺标本的刚果红阳性区/微解剖区提取的肽进行液相色谱-串联质谱分析。检测到的肽谱与AA (SAA)型淀粉样蛋白沉积一致。患者的基因检测结果显示E148Q纯合子突变。这证实了FMF的临床证候。持续随访10年后,患者发展为终末期肾衰竭,肾活检证实肾AA淀粉样变。从那以后,她一直在进行血液透析,现在正在等待肾脏移植。淀粉样甲状腺肿仅见于0.04%的全身性淀粉样变性患者淀粉样蛋白沉积可能是局部的,也可能是全身性的,正如本例患者,两者都有,但最初表现为局部甲状腺肿。国际淀粉样变性学会指南将淀粉样蛋白定义为一种纤维蛋白的细胞外沉积,通过其在偏光下对刚果红和苹果绿双折射的亲和力来识别我们证实了淀粉样蛋白沉积在我们的情况下,后者的辅助研究。迄今为止,有36种蛋白质被确定为人类淀粉样蛋白。AL是由免疫球蛋白轻链引起的淀粉样变,是发达国家最常见的系统性淀粉样变在我们的病例中,我们确定了aa型淀粉样蛋白沉积的一个原因,淀粉样蛋白原纤维来源于血清淀粉样蛋白a,这是发展中国家系统性淀粉样变性的主要原因AA蛋白与遗传性自身免疫性疾病有关,如本病例中的FMF。FMF具有常染色体隐性遗传和基因多态性,可导致淀粉样变性。这种实体是由于位于16号染色体上的地中海热基因(MEFV)的功能突变的获得(16p13.3)MEFV基因有10个外显子,迄今已确定的变体超过370个。FMF致病性最高的突变是外显子10 11的M694V、M694I、M680I和V726A,本病例未发现。在大多数FMF病例中,外显子2中的E148Q变异是MEFV基因中最常见的替换之一。它是唯一确定的变异或与其他变异平行发生,包括外显子10突变。有限的证据支持纯合子E148Q是一种致病突变。在我们的病人身上,这就是检测到的变异。外显子2的E148Q变异常见于日本FMF患者12,这可能与本患者的亚裔美国血统有关。根据文献,纯合子E148Q变异的患者可能表现为晚发性和轻度疾病13,14,尽管这些病例均未表现为淀粉样甲状腺肿。 淀粉样甲状腺肿的确切患病率尚未确定。Vergneault等人发现了15.00%,Ozdemir研究发现了15.45.00%,Altiparmak等人发现了0.27%。之前的报道都没有对患者进行基因检测。这些病例大多有其他全身性症状,最常见的是肾功能衰竭。我们的病人出现了预期的肾功能衰竭。孤立性淀粉样甲状腺肿是FMF的一种独特表现。尽管文献中有淀粉样甲状腺肿的报道,但大多数患者在出现疾病时都有其他全身性疾病。本病例的纯合子E148Q变体很有趣,因为它的疾病表现轻微,并且在文献中与淀粉样甲状腺肿缺乏关联。重要的是要认识到弥漫性甲状腺肿可能是FMF的早期临床表现。Juan C. A. Moreno研究了病例报告的概念,并为数据收集做出了贡献。Juan C. A. Moreno, Suimin Qiu和Eduardo Eyzaguirre参与病理切片回顾和数据分析。胡安·c·a·莫雷诺负责从医院的医疗记录中获取CT图像。Eduardo Eyzaguirre和Suimin Qiu对报告的病例进行了解释。Juan C. A. Moreno负责组织学图、图形插图和案例研究时间轴展示。邱敏敏负责学习监督。在最终定稿前,所有作者都对稿件进行了严格的修改和编辑。作者声明无利益冲突。这项工作是按照世界医学协会的道德守则(赫尔辛基宣言)进行的。
Amyloid goiter secondary to familial Mediterranean fever with E148Q mutation: A unique case
Dear Editor,
Goiter is defined as the enlargement of the thyroid gland. It is currently divided into diffuse and nodular and subdivided into toxic (associated with hyperthyroidism) or nontoxic (associated with normal thyroid stimulating hormone [TSH] levels).1 The most common cause of goiter is iodine deficiency,2 and other causes are increased levels of TSH, natural goitrogens, iron and vitamin A deficiency, genetic factors (DICER1 syndrome and PTEN hamartoma tumor syndrome), and hereditary (Plummer syndrome) factors.3
A rare entity known as familial Mediterranean fever (FMF) can present with amyloid goiter. This is an autoimmune disorder that affects the Mediterranean littoral.4 These patients present with fevers and abdominal and chest pain. This condition can produce fibrillar depositions of amyloid protein, usually affecting the kidney5 but rarely involving the thyroid. We present a case of a 21-year-old woman with no medical history who presented to our hospital with a nontoxic diffuse goiter with initial presentation and pathologically confirmed as amyloid deposition secondary to FMF.
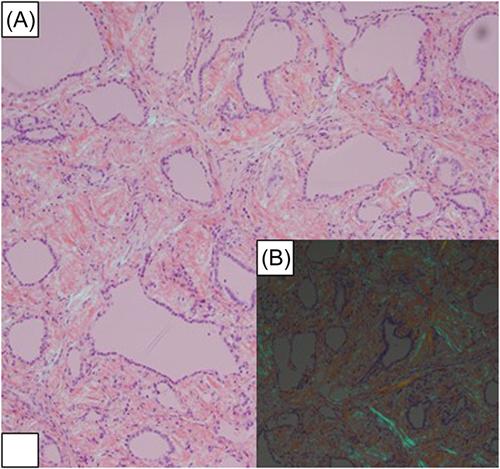
The patient is a 21-year-old Asian American woman with no significant medical history. She presented to our institution with dyspnea and dysphagia. An ultrasound from an outside hospital revealed diffuse thyroid enlargement. Our in-house laboratory results showed normal TSH, T4, T3, and elevated TPO antibody and erythrocyte sedimentation rate. A CT scan was performed on the patient showing heterogenous thyromegaly wrapping around the trachea and esophagus (Figure 1). A fine needle aspirate was performed, which showed a fibroinflammatory lesion. The patient started on steroids with no improvement, and a total thyroidectomy was performed. Macroscopic examination revealed a poorly defined, firm mass with pale tan areas of discoloration (Figure 2). Microscopic examination revealed an atrophic thyroid parenchyma with diffuse adipose cell metaplasia and diffuse interfollicular deposition of acellular amorphous material consistent with amyloid (Figure 3). Multifocal areas of chronic inflammation were also seen. Congo red stain was positive for amyloid (Figure 4A) with apple-green birefringence under polarized light (Figure 4B). Liquid chromatography tandem mass spectrometry was performed on peptides extracted from the Congo red-positive/microdissected areas of the paraffin-embedded thyroid specimen. The detected peptide profile was consistent with AA (SAA)-type amyloid deposition. The patient consequently had genetic testing showing a homozygous E148Q mutation. This confirms the clinical syndrome of FMF. After continuous follow-up, 10 years later, she developed end-stage renal failure with a renal biopsy confirming Renal AA amyloidosis. Since then, she has been on hemodialysis and is now on the waiting list for a renal transplant.
Amyloid goiter is only seen in 0.04% of patients with systemic amyloidosis.6 Amyloid depositions may be localized or systemic, as in our patient who developed both but initially presented as localized goiter. The International Society of Amyloidosis guidelines defines amyloid as an extracellular deposition of a fibrillary protein that is recognizable by its affinity for Congo red and its apple-green birefringence under polarized light.7 We confirm the amyloid depositions in our case with the latter ancillary studies.
To date, there are 36 proteins identified as amyloidogenic in humans. AL, which is amyloidosis derived from immunoglobulin light chain, is the most common presentation of systemic amyloidosis in the developed world.8 In our case, we identified a cause of AA-type amyloid deposition, amyloid fibrils derived from serum amyloid A protein, which is the leading cause of systemic amyloidosis in developing countries.9 The AA protein is associated with hereditary autoimmune diseases, such as, in our case, FMF.
FMF has autosomal recessive inheritance with gene polymorphism and can result in amyloidosis. This entity results from the gain of function mutations of the Mediterranean fever gene (MEFV) located on chromosome 16 (16p13.3).10 MEFV gene has 10 exons, and there are more than 370 variants identified to date. The most pathogenic mutations in FMF are M694V, M694I, M680I, and V726A in exon 10,11 which were not found in our case. The E148Q variant in exon 2 is one of the most common substitutions in the MEFV gene11 in most FMF cases. It occurs as the only identified variant or in parallel with other variants, including exon 10 mutations. Limited evidence supports that homozygous E148Q is a disease-causing mutation. In our patient, this was the variant detected. The E148Q variant in exon 2 is found frequently in Japanese patients with FMF,12 which possibly correlates with our patient's Asian American heritage. According to the literature, patients with homozygous E148Q variant may present with late-onset and mild disease,13, 14 although none of these cases presented with amyloid goiter. The exact prevalence of amyloid goiter in this entity is yet to be established. Approximately 15.00% was found by Vergneault et al.,15 45.00% in the Ozdemir study,16 and 0.27% in Altiparmak et al.17 findings. None of the previous reports had genetic testing on the patients. Most of these cases had other systemic symptoms, renal failure being the most common. Our patient developed renal failure in a prospective fashion.
This case of isolated amyloid goiter is a unique presentation of FMF. Despite the literature having reports of amyloid goiter, most of the patients had other systemic diseases at the time of presentation of the disease. The homozygous E148Q variant in this case is interesting, due to its mild disease presentation and lack of association with amyloid goiter in the literature. It is important to recognize that diffuse goiter can be the early clinical presentation of FMF.
Juan C. A. Moreno worked on the case report conception and contributed to the data collection. Juan C. A. Moreno, Suimin Qiu, and Eduardo Eyzaguirre contributed to the pathological slides review and data analysis. Juan C. A. Moreno was responsible for getting the CT images from the medical records of the hospital. Eduardo Eyzaguirre and Suimin Qiu provided explanations about the case reported. Juan C. A. Moreno worked on the histology figures, figure illustrations, and case study timeline presentation. Suimin Qiu was responsible for the study supervision. All authors critically revised and edited the manuscript before approving the final draft of the manuscript.
The authors declare no conflict of interest.
The work has been carried out in accordance with the code of ethics of the world medical association (Declaration of Helsinki).
期刊介绍:
This journal aims to promote progress from basic research to clinical practice and to provide a forum for communication among basic, translational, and clinical research practitioners and physicians from all relevant disciplines. Chronic diseases such as cardiovascular diseases, cancer, diabetes, stroke, chronic respiratory diseases (such as asthma and COPD), chronic kidney diseases, and related translational research. Topics of interest for Chronic Diseases and Translational Medicine include Research and commentary on models of chronic diseases with significant implications for disease diagnosis and treatment Investigative studies of human biology with an emphasis on disease Perspectives and reviews on research topics that discuss the implications of findings from the viewpoints of basic science and clinical practic.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
