Tahereh Ghorashi, Hossein Darvish, Somayeh Bakhtiari, Abbas Tafakhori, Michael C Kruer, Hossein Mozdarani
{"title":"TMEM147中的双等位基因功能缺失变体会导致严重的智力残疾和痉挛。","authors":"Tahereh Ghorashi, Hossein Darvish, Somayeh Bakhtiari, Abbas Tafakhori, Michael C Kruer, Hossein Mozdarani","doi":"10.1007/s10048-023-00734-8","DOIUrl":null,"url":null,"abstract":"<p><p>Intellectual disability (ID), occurring in syndromic or non-syndromic forms, is the most common neurodevelopmental disorder. Although many cases are caused by single gene defects, ID is highly genetically heterogeneous. Biallelic variants in the transmembrane protein TMEM147 have recently been linked to intellectual disability with dysmorphic facial features. TMEM147 is believed to localize to the endoplasmic reticulum membrane and nuclear envelope and also involved in biogenesis of multi-pass membrane proteins. Here, we report two patients born to a consanguineous family with a novel loss-of-function variant; (NM_001242597.2:c.193-197del) in TMEM147 causing intellectual disability and spasticity. Whole exome sequencing and validating Sanger sequencing were utilized to confirm the identified causal variant. Our findings were in line with the previously described patients with TMEM147 variants manifesting intellectual disability as a major clinical sign but also featured spasticity as a phenotypic expansion. This study provides additional evidence for the pathogenicity of TMEM147 mutations in intellectual disability and expands the phenotypic and variant spectrum linked to this gene.</p>","PeriodicalId":56106,"journal":{"name":"Neurogenetics","volume":" ","pages":"311-316"},"PeriodicalIF":1.6000,"publicationDate":"2023-10-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"A biallelic loss-of-function variant in TMEM147 causes profound intellectual disability and spasticity.\",\"authors\":\"Tahereh Ghorashi, Hossein Darvish, Somayeh Bakhtiari, Abbas Tafakhori, Michael C Kruer, Hossein Mozdarani\",\"doi\":\"10.1007/s10048-023-00734-8\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Intellectual disability (ID), occurring in syndromic or non-syndromic forms, is the most common neurodevelopmental disorder. Although many cases are caused by single gene defects, ID is highly genetically heterogeneous. Biallelic variants in the transmembrane protein TMEM147 have recently been linked to intellectual disability with dysmorphic facial features. TMEM147 is believed to localize to the endoplasmic reticulum membrane and nuclear envelope and also involved in biogenesis of multi-pass membrane proteins. Here, we report two patients born to a consanguineous family with a novel loss-of-function variant; (NM_001242597.2:c.193-197del) in TMEM147 causing intellectual disability and spasticity. Whole exome sequencing and validating Sanger sequencing were utilized to confirm the identified causal variant. Our findings were in line with the previously described patients with TMEM147 variants manifesting intellectual disability as a major clinical sign but also featured spasticity as a phenotypic expansion. This study provides additional evidence for the pathogenicity of TMEM147 mutations in intellectual disability and expands the phenotypic and variant spectrum linked to this gene.</p>\",\"PeriodicalId\":56106,\"journal\":{\"name\":\"Neurogenetics\",\"volume\":\" \",\"pages\":\"311-316\"},\"PeriodicalIF\":1.6000,\"publicationDate\":\"2023-10-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Neurogenetics\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://doi.org/10.1007/s10048-023-00734-8\",\"RegionNum\":4,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2023/9/5 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"Q3\",\"JCRName\":\"CLINICAL NEUROLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Neurogenetics","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1007/s10048-023-00734-8","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2023/9/5 0:00:00","PubModel":"Epub","JCR":"Q3","JCRName":"CLINICAL NEUROLOGY","Score":null,"Total":0}
A biallelic loss-of-function variant in TMEM147 causes profound intellectual disability and spasticity.
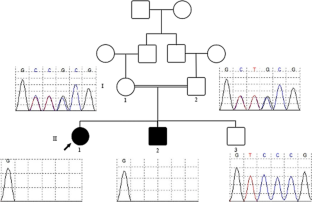
Intellectual disability (ID), occurring in syndromic or non-syndromic forms, is the most common neurodevelopmental disorder. Although many cases are caused by single gene defects, ID is highly genetically heterogeneous. Biallelic variants in the transmembrane protein TMEM147 have recently been linked to intellectual disability with dysmorphic facial features. TMEM147 is believed to localize to the endoplasmic reticulum membrane and nuclear envelope and also involved in biogenesis of multi-pass membrane proteins. Here, we report two patients born to a consanguineous family with a novel loss-of-function variant; (NM_001242597.2:c.193-197del) in TMEM147 causing intellectual disability and spasticity. Whole exome sequencing and validating Sanger sequencing were utilized to confirm the identified causal variant. Our findings were in line with the previously described patients with TMEM147 variants manifesting intellectual disability as a major clinical sign but also featured spasticity as a phenotypic expansion. This study provides additional evidence for the pathogenicity of TMEM147 mutations in intellectual disability and expands the phenotypic and variant spectrum linked to this gene.
期刊介绍:
Neurogenetics publishes findings that contribute to a better understanding of the genetic basis of normal and abnormal function of the nervous system. Neurogenetic disorders are the main focus of the journal. Neurogenetics therefore includes findings in humans and other organisms that help understand neurological disease mechanisms and publishes papers from many different fields such as biophysics, cell biology, human genetics, neuroanatomy, neurochemistry, neurology, neuropathology, neurosurgery and psychiatry.
All papers submitted to Neurogenetics should be of sufficient immediate importance to justify urgent publication. They should present new scientific results. Data merely confirming previously published findings are not acceptable.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
