Jie Wang, Zili Zuo, Zongze Yu, Zhigui Chen, Xiangdi Meng, Zhaosen Ma, Jiqiang Niu, Rui Guo, Lisa Jia Tran, Jing Zhang, Tianxiao Jiang, Fangdie Ye, Baoluo Ma, Zhou Sun
{"title":"单细胞转录组分析揭示了ccRCC的肿瘤内异质性和MT2A在发病机制中的验证","authors":"Jie Wang, Zili Zuo, Zongze Yu, Zhigui Chen, Xiangdi Meng, Zhaosen Ma, Jiqiang Niu, Rui Guo, Lisa Jia Tran, Jing Zhang, Tianxiao Jiang, Fangdie Ye, Baoluo Ma, Zhou Sun","doi":"10.1007/s10142-023-01225-7","DOIUrl":null,"url":null,"abstract":"<div><p>Clear-cell renal cell carcinoma (ccRCC) appears as the most common type of kidney cancer, the carcinogenesis of which has not been fully elucidated. Tumor heterogeneity plays a crucial role in cancer progression, which could be largely deciphered by the implement of scRNA-seq. The bulk and single-cell RNA expression profile is obtained from TCGA and study conducted by Young et al. We utilized UMAP, TSNE, and clustering algorithm Louvain for dimensionality reduction and FindAllMarkers function for determining the DEGs. Monocle2 was utilized to perform pseudo-time series analysis. SCENIC was implemented for transcription factor analysis of each cell subgroup. A series of WB, CFA, CCK-8, and EDU analysis was utilized for the validation of the role of MT2A in ccRCC carcinogenesis. We observed higher infiltration of T/NK and B cells in tumorous tissues, indicating the role of immune cells in ccRCC carcinogenesis. Transcription factor analysis revealed the activation of EOMES and ETS1 in CD8 + T cells, while CAFs were divided into myo-CAFs and i-CAFs, with i-CAFs showing distinct enrichment of ATF3, JUND, JUNB, EGR1, and XBP1. Through cell trajectory analysis, we discerned three distinct stages of cellular evolution, where State2 symbolizes normal renal tubular cells that underwent transitions into State1 and State3 as the CNV score ascended. Functional enrichment examination revealed an amplification of interferon gamma and inflammatory response pathways within tumor cells. The consensus clustering algorithm yielded two molecular subtypes, with cluster 2 being associated with advanced tumor stages and an abundance of infiltrated immune cells. We identified 17 prognostic genes through Cox and LASSO regression models and used them to construct a prognostic model, the efficacy of which was verified in multiple cohorts. Furthermore, we investigated the role of MT2A, one of our hub genes, in ccRCC carcinogenesis, and found it to regulate proliferation and migration of malignant cells. We depicted a detailed single-cell landscape of ccRCC, with special focus on CAFs, endothelial cells, and renal tubular cells. A prognostic model of high stability and accuracy was constructed based on the DEGs. MT2A was found to be actively implicated in ccRCC carcinogenesis, regulating proliferation and migration of the malignant cells.</p></div>","PeriodicalId":574,"journal":{"name":"Functional & Integrative Genomics","volume":"23 4","pages":""},"PeriodicalIF":3.1000,"publicationDate":"2023-09-15","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"3","resultStr":"{\"title\":\"Single-cell transcriptome analysis revealing the intratumoral heterogeneity of ccRCC and validation of MT2A in pathogenesis\",\"authors\":\"Jie Wang, Zili Zuo, Zongze Yu, Zhigui Chen, Xiangdi Meng, Zhaosen Ma, Jiqiang Niu, Rui Guo, Lisa Jia Tran, Jing Zhang, Tianxiao Jiang, Fangdie Ye, Baoluo Ma, Zhou Sun\",\"doi\":\"10.1007/s10142-023-01225-7\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<div><p>Clear-cell renal cell carcinoma (ccRCC) appears as the most common type of kidney cancer, the carcinogenesis of which has not been fully elucidated. Tumor heterogeneity plays a crucial role in cancer progression, which could be largely deciphered by the implement of scRNA-seq. The bulk and single-cell RNA expression profile is obtained from TCGA and study conducted by Young et al. We utilized UMAP, TSNE, and clustering algorithm Louvain for dimensionality reduction and FindAllMarkers function for determining the DEGs. Monocle2 was utilized to perform pseudo-time series analysis. SCENIC was implemented for transcription factor analysis of each cell subgroup. A series of WB, CFA, CCK-8, and EDU analysis was utilized for the validation of the role of MT2A in ccRCC carcinogenesis. We observed higher infiltration of T/NK and B cells in tumorous tissues, indicating the role of immune cells in ccRCC carcinogenesis. Transcription factor analysis revealed the activation of EOMES and ETS1 in CD8 + T cells, while CAFs were divided into myo-CAFs and i-CAFs, with i-CAFs showing distinct enrichment of ATF3, JUND, JUNB, EGR1, and XBP1. Through cell trajectory analysis, we discerned three distinct stages of cellular evolution, where State2 symbolizes normal renal tubular cells that underwent transitions into State1 and State3 as the CNV score ascended. Functional enrichment examination revealed an amplification of interferon gamma and inflammatory response pathways within tumor cells. The consensus clustering algorithm yielded two molecular subtypes, with cluster 2 being associated with advanced tumor stages and an abundance of infiltrated immune cells. We identified 17 prognostic genes through Cox and LASSO regression models and used them to construct a prognostic model, the efficacy of which was verified in multiple cohorts. Furthermore, we investigated the role of MT2A, one of our hub genes, in ccRCC carcinogenesis, and found it to regulate proliferation and migration of malignant cells. We depicted a detailed single-cell landscape of ccRCC, with special focus on CAFs, endothelial cells, and renal tubular cells. A prognostic model of high stability and accuracy was constructed based on the DEGs. MT2A was found to be actively implicated in ccRCC carcinogenesis, regulating proliferation and migration of the malignant cells.</p></div>\",\"PeriodicalId\":574,\"journal\":{\"name\":\"Functional & Integrative Genomics\",\"volume\":\"23 4\",\"pages\":\"\"},\"PeriodicalIF\":3.1000,\"publicationDate\":\"2023-09-15\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"3\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Functional & Integrative Genomics\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://link.springer.com/article/10.1007/s10142-023-01225-7\",\"RegionNum\":4,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"GENETICS & HEREDITY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Functional & Integrative Genomics","FirstCategoryId":"99","ListUrlMain":"https://link.springer.com/article/10.1007/s10142-023-01225-7","RegionNum":4,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
Single-cell transcriptome analysis revealing the intratumoral heterogeneity of ccRCC and validation of MT2A in pathogenesis
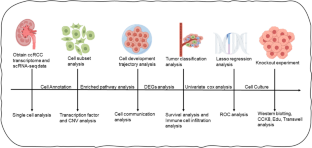
Clear-cell renal cell carcinoma (ccRCC) appears as the most common type of kidney cancer, the carcinogenesis of which has not been fully elucidated. Tumor heterogeneity plays a crucial role in cancer progression, which could be largely deciphered by the implement of scRNA-seq. The bulk and single-cell RNA expression profile is obtained from TCGA and study conducted by Young et al. We utilized UMAP, TSNE, and clustering algorithm Louvain for dimensionality reduction and FindAllMarkers function for determining the DEGs. Monocle2 was utilized to perform pseudo-time series analysis. SCENIC was implemented for transcription factor analysis of each cell subgroup. A series of WB, CFA, CCK-8, and EDU analysis was utilized for the validation of the role of MT2A in ccRCC carcinogenesis. We observed higher infiltration of T/NK and B cells in tumorous tissues, indicating the role of immune cells in ccRCC carcinogenesis. Transcription factor analysis revealed the activation of EOMES and ETS1 in CD8 + T cells, while CAFs were divided into myo-CAFs and i-CAFs, with i-CAFs showing distinct enrichment of ATF3, JUND, JUNB, EGR1, and XBP1. Through cell trajectory analysis, we discerned three distinct stages of cellular evolution, where State2 symbolizes normal renal tubular cells that underwent transitions into State1 and State3 as the CNV score ascended. Functional enrichment examination revealed an amplification of interferon gamma and inflammatory response pathways within tumor cells. The consensus clustering algorithm yielded two molecular subtypes, with cluster 2 being associated with advanced tumor stages and an abundance of infiltrated immune cells. We identified 17 prognostic genes through Cox and LASSO regression models and used them to construct a prognostic model, the efficacy of which was verified in multiple cohorts. Furthermore, we investigated the role of MT2A, one of our hub genes, in ccRCC carcinogenesis, and found it to regulate proliferation and migration of malignant cells. We depicted a detailed single-cell landscape of ccRCC, with special focus on CAFs, endothelial cells, and renal tubular cells. A prognostic model of high stability and accuracy was constructed based on the DEGs. MT2A was found to be actively implicated in ccRCC carcinogenesis, regulating proliferation and migration of the malignant cells.
期刊介绍:
Functional & Integrative Genomics is devoted to large-scale studies of genomes and their functions, including systems analyses of biological processes. The journal will provide the research community an integrated platform where researchers can share, review and discuss their findings on important biological questions that will ultimately enable us to answer the fundamental question: How do genomes work?


 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
