Nica Borgese, Francesca Navone, Nobuyuki Nukina, Tomoyuki Yamanaka
{"title":"变异的VAPB:肌萎缩性侧索硬化症的罪魁祸首还是无辜的旁观者?","authors":"Nica Borgese, Francesca Navone, Nobuyuki Nukina, Tomoyuki Yamanaka","doi":"10.1177/25152564211022515","DOIUrl":null,"url":null,"abstract":"<p><p>Nearly twenty years ago a mutation in the VAPB gene, resulting in a proline to serine substitution (p.P56S), was identified as the cause of a rare, slowly progressing, familial form of the motor neuron degenerative disease Amyotrophic Lateral Sclerosis (ALS). Since then, progress in unravelling the mechanistic basis of this mutation has proceeded in parallel with research on the VAP proteins and on their role in establishing membrane contact sites between the ER and other organelles. Analysis of the literature on cellular and animal models reviewed here supports the conclusion that P56S-VAPB, which is aggregation-prone, non-functional and unstable, is expressed at levels that are insufficient to support toxic gain-of-function or dominant negative effects within motor neurons. Instead, insufficient levels of the product of the single wild-type allele appear to be required for pathological effects, and may be the main driver of the disease. In light of the multiple interactions of the VAP proteins, we address the consequences of specific VAPB depletion and highlight various affected processes that could contribute to motor neuron degeneration. In the future, distinction of specific roles of each of the two VAP paralogues should help to further elucidate the basis of p.P56S familial ALS, as well as of other more common forms of the disease.</p>","PeriodicalId":10556,"journal":{"name":"Contact (Thousand Oaks (Ventura County, Calif.))","volume":"4 ","pages":"25152564211022515"},"PeriodicalIF":0.0000,"publicationDate":"2021-01-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.1177/25152564211022515","citationCount":"6","resultStr":"{\"title\":\"Mutant VAPB: Culprit or Innocent Bystander of Amyotrophic Lateral Sclerosis?\",\"authors\":\"Nica Borgese, Francesca Navone, Nobuyuki Nukina, Tomoyuki Yamanaka\",\"doi\":\"10.1177/25152564211022515\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Nearly twenty years ago a mutation in the VAPB gene, resulting in a proline to serine substitution (p.P56S), was identified as the cause of a rare, slowly progressing, familial form of the motor neuron degenerative disease Amyotrophic Lateral Sclerosis (ALS). Since then, progress in unravelling the mechanistic basis of this mutation has proceeded in parallel with research on the VAP proteins and on their role in establishing membrane contact sites between the ER and other organelles. Analysis of the literature on cellular and animal models reviewed here supports the conclusion that P56S-VAPB, which is aggregation-prone, non-functional and unstable, is expressed at levels that are insufficient to support toxic gain-of-function or dominant negative effects within motor neurons. Instead, insufficient levels of the product of the single wild-type allele appear to be required for pathological effects, and may be the main driver of the disease. In light of the multiple interactions of the VAP proteins, we address the consequences of specific VAPB depletion and highlight various affected processes that could contribute to motor neuron degeneration. In the future, distinction of specific roles of each of the two VAP paralogues should help to further elucidate the basis of p.P56S familial ALS, as well as of other more common forms of the disease.</p>\",\"PeriodicalId\":10556,\"journal\":{\"name\":\"Contact (Thousand Oaks (Ventura County, Calif.))\",\"volume\":\"4 \",\"pages\":\"25152564211022515\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2021-01-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://sci-hub-pdf.com/10.1177/25152564211022515\",\"citationCount\":\"6\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Contact (Thousand Oaks (Ventura County, Calif.))\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1177/25152564211022515\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"\",\"JCRName\":\"\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Contact (Thousand Oaks (Ventura County, Calif.))","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1177/25152564211022515","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"","JCRName":"","Score":null,"Total":0}
Mutant VAPB: Culprit or Innocent Bystander of Amyotrophic Lateral Sclerosis?
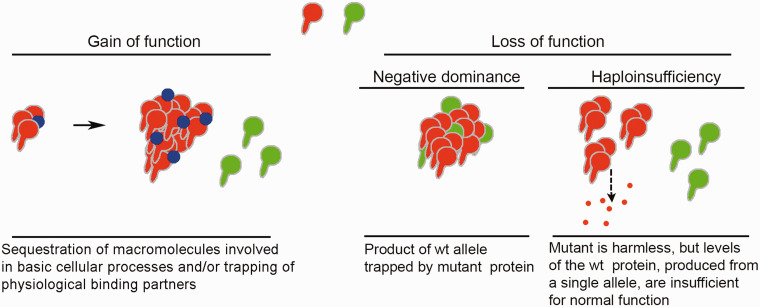
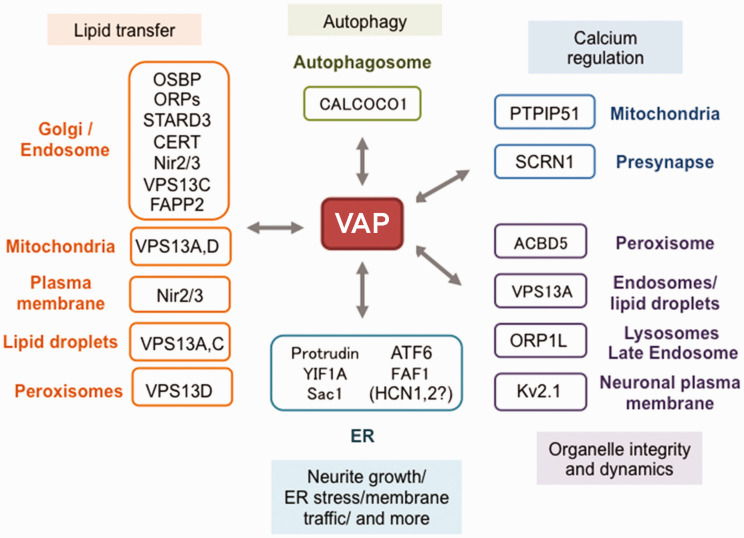
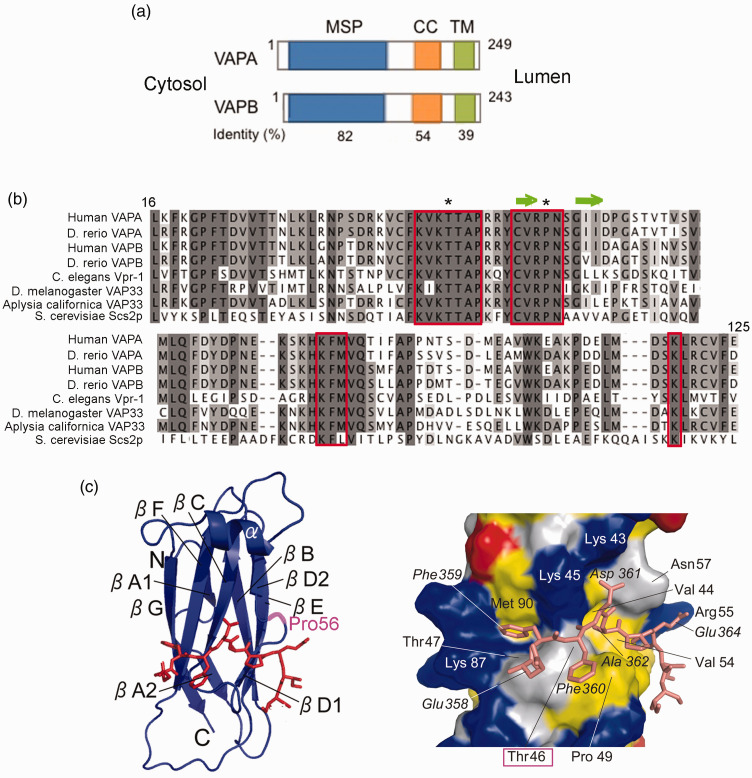
Nearly twenty years ago a mutation in the VAPB gene, resulting in a proline to serine substitution (p.P56S), was identified as the cause of a rare, slowly progressing, familial form of the motor neuron degenerative disease Amyotrophic Lateral Sclerosis (ALS). Since then, progress in unravelling the mechanistic basis of this mutation has proceeded in parallel with research on the VAP proteins and on their role in establishing membrane contact sites between the ER and other organelles. Analysis of the literature on cellular and animal models reviewed here supports the conclusion that P56S-VAPB, which is aggregation-prone, non-functional and unstable, is expressed at levels that are insufficient to support toxic gain-of-function or dominant negative effects within motor neurons. Instead, insufficient levels of the product of the single wild-type allele appear to be required for pathological effects, and may be the main driver of the disease. In light of the multiple interactions of the VAP proteins, we address the consequences of specific VAPB depletion and highlight various affected processes that could contribute to motor neuron degeneration. In the future, distinction of specific roles of each of the two VAP paralogues should help to further elucidate the basis of p.P56S familial ALS, as well as of other more common forms of the disease.



 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
