Yongjun Liu, Heping Zhang, Yuqing Xu, Yao-Zhong Liu, David P Al-Adra, Matthew M Yeh, Zhengjun Zhang
{"title":"五种关键基因生物标志物在肝细胞癌中的最佳表现。","authors":"Yongjun Liu, Heping Zhang, Yuqing Xu, Yao-Zhong Liu, David P Al-Adra, Matthew M Yeh, Zhengjun Zhang","doi":"10.1177/11769351231190477","DOIUrl":null,"url":null,"abstract":"Hepatocellular carcinoma (HCC) is one of the most fatal cancers in the world. There is an urgent need to understand the molecular background of HCC to facilitate the identification of biomarkers and discover effective therapeutic targets. Published transcriptomic studies have reported a large number of genes that are individually significant for HCC. However, reliable biomarkers remain to be determined. In this study, built on max-linear competing risk factor models, we developed a machine learning analytical framework to analyze transcriptomic data to identify the most miniature set of differentially expressed genes (DEGs). By analyzing 9 public whole-transcriptome datasets (containing 1184 HCC samples and 672 nontumor controls), we identified 5 critical differentially expressed genes (DEGs) (ie, CCDC107, CXCL12, GIGYF1, GMNN, and IFFO1) between HCC and control samples. The classifiers built on these 5 DEGs reached nearly perfect performance in identification of HCC. The performance of the 5 DEGs was further validated in a US Caucasian cohort that we collected (containing 17 HCC with paired nontumor tissue). The conceptual advance of our work lies in modeling gene-gene interactions and correcting batch effect in the analytic framework. The classifiers built on the 5 DEGs demonstrated clear signature patterns for HCC. The results are interpretable, robust, and reproducible across diverse cohorts/populations with various disease etiologies, indicating the 5 DEGs are intrinsic variables that can describe the overall features of HCC at the genomic level. The analytical framework applied in this study may pave a new way for improving transcriptome profiling analysis of human cancers.","PeriodicalId":35418,"journal":{"name":"Cancer Informatics","volume":"22 ","pages":"11769351231190477"},"PeriodicalIF":2.5000,"publicationDate":"2023-01-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/11/97/10.1177_11769351231190477.PMC10413891.pdf","citationCount":"0","resultStr":"{\"title\":\"Five Critical Gene-Based Biomarkers With Optimal Performance for Hepatocellular Carcinoma.\",\"authors\":\"Yongjun Liu, Heping Zhang, Yuqing Xu, Yao-Zhong Liu, David P Al-Adra, Matthew M Yeh, Zhengjun Zhang\",\"doi\":\"10.1177/11769351231190477\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"Hepatocellular carcinoma (HCC) is one of the most fatal cancers in the world. There is an urgent need to understand the molecular background of HCC to facilitate the identification of biomarkers and discover effective therapeutic targets. Published transcriptomic studies have reported a large number of genes that are individually significant for HCC. However, reliable biomarkers remain to be determined. In this study, built on max-linear competing risk factor models, we developed a machine learning analytical framework to analyze transcriptomic data to identify the most miniature set of differentially expressed genes (DEGs). By analyzing 9 public whole-transcriptome datasets (containing 1184 HCC samples and 672 nontumor controls), we identified 5 critical differentially expressed genes (DEGs) (ie, CCDC107, CXCL12, GIGYF1, GMNN, and IFFO1) between HCC and control samples. The classifiers built on these 5 DEGs reached nearly perfect performance in identification of HCC. The performance of the 5 DEGs was further validated in a US Caucasian cohort that we collected (containing 17 HCC with paired nontumor tissue). The conceptual advance of our work lies in modeling gene-gene interactions and correcting batch effect in the analytic framework. The classifiers built on the 5 DEGs demonstrated clear signature patterns for HCC. The results are interpretable, robust, and reproducible across diverse cohorts/populations with various disease etiologies, indicating the 5 DEGs are intrinsic variables that can describe the overall features of HCC at the genomic level. The analytical framework applied in this study may pave a new way for improving transcriptome profiling analysis of human cancers.\",\"PeriodicalId\":35418,\"journal\":{\"name\":\"Cancer Informatics\",\"volume\":\"22 \",\"pages\":\"11769351231190477\"},\"PeriodicalIF\":2.5000,\"publicationDate\":\"2023-01-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/11/97/10.1177_11769351231190477.PMC10413891.pdf\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Cancer Informatics\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1177/11769351231190477\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"MATHEMATICAL & COMPUTATIONAL BIOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Cancer Informatics","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1177/11769351231190477","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"MATHEMATICAL & COMPUTATIONAL BIOLOGY","Score":null,"Total":0}
Five Critical Gene-Based Biomarkers With Optimal Performance for Hepatocellular Carcinoma.
Hepatocellular carcinoma (HCC) is one of the most fatal cancers in the world. There is an urgent need to understand the molecular background of HCC to facilitate the identification of biomarkers and discover effective therapeutic targets. Published transcriptomic studies have reported a large number of genes that are individually significant for HCC. However, reliable biomarkers remain to be determined. In this study, built on max-linear competing risk factor models, we developed a machine learning analytical framework to analyze transcriptomic data to identify the most miniature set of differentially expressed genes (DEGs). By analyzing 9 public whole-transcriptome datasets (containing 1184 HCC samples and 672 nontumor controls), we identified 5 critical differentially expressed genes (DEGs) (ie, CCDC107, CXCL12, GIGYF1, GMNN, and IFFO1) between HCC and control samples. The classifiers built on these 5 DEGs reached nearly perfect performance in identification of HCC. The performance of the 5 DEGs was further validated in a US Caucasian cohort that we collected (containing 17 HCC with paired nontumor tissue). The conceptual advance of our work lies in modeling gene-gene interactions and correcting batch effect in the analytic framework. The classifiers built on the 5 DEGs demonstrated clear signature patterns for HCC. The results are interpretable, robust, and reproducible across diverse cohorts/populations with various disease etiologies, indicating the 5 DEGs are intrinsic variables that can describe the overall features of HCC at the genomic level. The analytical framework applied in this study may pave a new way for improving transcriptome profiling analysis of human cancers.
期刊介绍:
The field of cancer research relies on advances in many other disciplines, including omics technology, mass spectrometry, radio imaging, computer science, and biostatistics. Cancer Informatics provides open access to peer-reviewed high-quality manuscripts reporting bioinformatics analysis of molecular genetics and/or clinical data pertaining to cancer, emphasizing the use of machine learning, artificial intelligence, statistical algorithms, advanced imaging techniques, data visualization, and high-throughput technologies. As the leading journal dedicated exclusively to the report of the use of computational methods in cancer research and practice, Cancer Informatics leverages methodological improvements in systems biology, genomics, proteomics, metabolomics, and molecular biochemistry into the fields of cancer detection, treatment, classification, risk-prediction, prevention, outcome, and modeling.
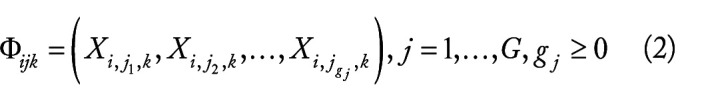
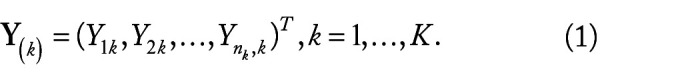
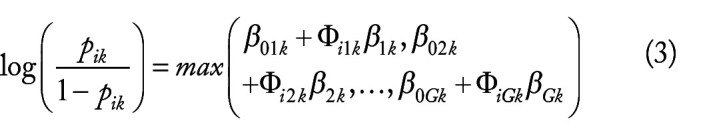




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
