Elias Vera-Siguenza, Cristina Escribano-Gonzalez, Irene Serrano-Gonzalo, Kattri-Liis Eskla, Fabian Spill, Daniel Tennant
{"title":"体外多发性骨髓瘤模型中代谢网络的数学重建。","authors":"Elias Vera-Siguenza, Cristina Escribano-Gonzalez, Irene Serrano-Gonzalo, Kattri-Liis Eskla, Fabian Spill, Daniel Tennant","doi":"10.1371/journal.pcbi.1011374","DOIUrl":null,"url":null,"abstract":"<p><p>It is increasingly apparent that cancer cells, in addition to remodelling their metabolism to survive and proliferate, adapt and manipulate the metabolism of other cells. This property may be a telling sign that pre-clinical tumour metabolism studies exclusively utilising in-vitro mono-culture models could prove to be limited for uncovering novel metabolic targets able to translate into clinical therapies. Although this is increasingly recognised, and work towards addressing the issue is becoming routinary much remains poorly understood. For instance, knowledge regarding the biochemical mechanisms through which cancer cells manipulate non-cancerous cell metabolism, and the subsequent impact on their survival and proliferation remains limited. Additionally, the variations in these processes across different cancer types and progression stages, and their implications for therapy, also remain largely unexplored. This study employs an interdisciplinary approach that leverages the predictive power of mathematical modelling to enrich experimental findings. We develop a functional multicellular in-silico model that facilitates the qualitative and quantitative analysis of the metabolic network spawned by an in-vitro co-culture model of bone marrow mesenchymal stem- and myeloma cell lines. To procure this model, we devised a bespoke human genome constraint-based reconstruction workflow that combines aspects from the legacy mCADRE & Metabotools algorithms, the novel redHuman algorithm, along with 13C-metabolic flux analysis. Our workflow transforms the latest human metabolic network matrix (Recon3D) into two cell-specific models coupled with a metabolic network spanning a shared growth medium. When cross-validating our in-silico model against the in-vitro model, we found that the in-silico model successfully reproduces vital metabolic behaviours of its in-vitro counterpart; results include cell growth predictions, respiration rates, as well as support for observations which suggest cross-shuttling of redox-active metabolites between cells.</p>","PeriodicalId":49688,"journal":{"name":"PLoS Computational Biology","volume":"19 9","pages":"e1011374"},"PeriodicalIF":3.6000,"publicationDate":"2023-09-15","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10503963/pdf/","citationCount":"0","resultStr":"{\"title\":\"Mathematical reconstruction of the metabolic network in an in-vitro multiple myeloma model.\",\"authors\":\"Elias Vera-Siguenza, Cristina Escribano-Gonzalez, Irene Serrano-Gonzalo, Kattri-Liis Eskla, Fabian Spill, Daniel Tennant\",\"doi\":\"10.1371/journal.pcbi.1011374\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>It is increasingly apparent that cancer cells, in addition to remodelling their metabolism to survive and proliferate, adapt and manipulate the metabolism of other cells. This property may be a telling sign that pre-clinical tumour metabolism studies exclusively utilising in-vitro mono-culture models could prove to be limited for uncovering novel metabolic targets able to translate into clinical therapies. Although this is increasingly recognised, and work towards addressing the issue is becoming routinary much remains poorly understood. For instance, knowledge regarding the biochemical mechanisms through which cancer cells manipulate non-cancerous cell metabolism, and the subsequent impact on their survival and proliferation remains limited. Additionally, the variations in these processes across different cancer types and progression stages, and their implications for therapy, also remain largely unexplored. This study employs an interdisciplinary approach that leverages the predictive power of mathematical modelling to enrich experimental findings. We develop a functional multicellular in-silico model that facilitates the qualitative and quantitative analysis of the metabolic network spawned by an in-vitro co-culture model of bone marrow mesenchymal stem- and myeloma cell lines. To procure this model, we devised a bespoke human genome constraint-based reconstruction workflow that combines aspects from the legacy mCADRE & Metabotools algorithms, the novel redHuman algorithm, along with 13C-metabolic flux analysis. Our workflow transforms the latest human metabolic network matrix (Recon3D) into two cell-specific models coupled with a metabolic network spanning a shared growth medium. When cross-validating our in-silico model against the in-vitro model, we found that the in-silico model successfully reproduces vital metabolic behaviours of its in-vitro counterpart; results include cell growth predictions, respiration rates, as well as support for observations which suggest cross-shuttling of redox-active metabolites between cells.</p>\",\"PeriodicalId\":49688,\"journal\":{\"name\":\"PLoS Computational Biology\",\"volume\":\"19 9\",\"pages\":\"e1011374\"},\"PeriodicalIF\":3.6000,\"publicationDate\":\"2023-09-15\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10503963/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"PLoS Computational Biology\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://doi.org/10.1371/journal.pcbi.1011374\",\"RegionNum\":2,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2023/9/1 0:00:00\",\"PubModel\":\"eCollection\",\"JCR\":\"\",\"JCRName\":\"\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"PLoS Computational Biology","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1371/journal.pcbi.1011374","RegionNum":2,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2023/9/1 0:00:00","PubModel":"eCollection","JCR":"","JCRName":"","Score":null,"Total":0}
Mathematical reconstruction of the metabolic network in an in-vitro multiple myeloma model.
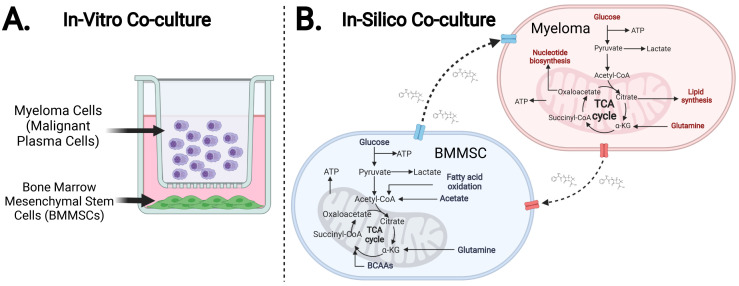
It is increasingly apparent that cancer cells, in addition to remodelling their metabolism to survive and proliferate, adapt and manipulate the metabolism of other cells. This property may be a telling sign that pre-clinical tumour metabolism studies exclusively utilising in-vitro mono-culture models could prove to be limited for uncovering novel metabolic targets able to translate into clinical therapies. Although this is increasingly recognised, and work towards addressing the issue is becoming routinary much remains poorly understood. For instance, knowledge regarding the biochemical mechanisms through which cancer cells manipulate non-cancerous cell metabolism, and the subsequent impact on their survival and proliferation remains limited. Additionally, the variations in these processes across different cancer types and progression stages, and their implications for therapy, also remain largely unexplored. This study employs an interdisciplinary approach that leverages the predictive power of mathematical modelling to enrich experimental findings. We develop a functional multicellular in-silico model that facilitates the qualitative and quantitative analysis of the metabolic network spawned by an in-vitro co-culture model of bone marrow mesenchymal stem- and myeloma cell lines. To procure this model, we devised a bespoke human genome constraint-based reconstruction workflow that combines aspects from the legacy mCADRE & Metabotools algorithms, the novel redHuman algorithm, along with 13C-metabolic flux analysis. Our workflow transforms the latest human metabolic network matrix (Recon3D) into two cell-specific models coupled with a metabolic network spanning a shared growth medium. When cross-validating our in-silico model against the in-vitro model, we found that the in-silico model successfully reproduces vital metabolic behaviours of its in-vitro counterpart; results include cell growth predictions, respiration rates, as well as support for observations which suggest cross-shuttling of redox-active metabolites between cells.
期刊介绍:
PLOS Computational Biology features works of exceptional significance that further our understanding of living systems at all scales—from molecules and cells, to patient populations and ecosystems—through the application of computational methods. Readers include life and computational scientists, who can take the important findings presented here to the next level of discovery.
Research articles must be declared as belonging to a relevant section. More information about the sections can be found in the submission guidelines.
Research articles should model aspects of biological systems, demonstrate both methodological and scientific novelty, and provide profound new biological insights.
Generally, reliability and significance of biological discovery through computation should be validated and enriched by experimental studies. Inclusion of experimental validation is not required for publication, but should be referenced where possible. Inclusion of experimental validation of a modest biological discovery through computation does not render a manuscript suitable for PLOS Computational Biology.
Research articles specifically designated as Methods papers should describe outstanding methods of exceptional importance that have been shown, or have the promise to provide new biological insights. The method must already be widely adopted, or have the promise of wide adoption by a broad community of users. Enhancements to existing published methods will only be considered if those enhancements bring exceptional new capabilities.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
