Eyal Simonovsky, Moran Sharon, Maya Ziv, Omry Mauer, Idan Hekselman, Juman Jubran, Ekaterina Vinogradov, Chanan M Argov, Omer Basha, Lior Kerber, Yuval Yogev, Ayellet V Segrè, Hae Kyung Im, Ohad Birk, Lior Rokach, Esti Yeger-Lotem
{"title":"利用遗传疾病的组织选择性表现预测遗传疾病的分子机制。","authors":"Eyal Simonovsky, Moran Sharon, Maya Ziv, Omry Mauer, Idan Hekselman, Juman Jubran, Ekaterina Vinogradov, Chanan M Argov, Omer Basha, Lior Kerber, Yuval Yogev, Ayellet V Segrè, Hae Kyung Im, Ohad Birk, Lior Rokach, Esti Yeger-Lotem","doi":"10.15252/msb.202211407","DOIUrl":null,"url":null,"abstract":"<p><p>How do aberrations in widely expressed genes lead to tissue-selective hereditary diseases? Previous attempts to answer this question were limited to testing a few candidate mechanisms. To answer this question at a larger scale, we developed \"Tissue Risk Assessment of Causality by Expression\" (TRACE), a machine learning approach to predict genes that underlie tissue-selective diseases and selectivity-related features. TRACE utilized 4,744 biologically interpretable tissue-specific gene features that were inferred from heterogeneous omics datasets. Application of TRACE to 1,031 disease genes uncovered known and novel selectivity-related features, the most common of which was previously overlooked. Next, we created a catalog of tissue-associated risks for 18,927 protein-coding genes (https://netbio.bgu.ac.il/trace/). As proof-of-concept, we prioritized candidate disease genes identified in 48 rare-disease patients. TRACE ranked the verified disease gene among the patient's candidate genes significantly better than gene prioritization methods that rank by gene constraint or tissue expression. Thus, tissue selectivity combined with machine learning enhances genetic and clinical understanding of hereditary diseases.</p>","PeriodicalId":18906,"journal":{"name":"Molecular Systems Biology","volume":"19 8","pages":"e11407"},"PeriodicalIF":7.7000,"publicationDate":"2023-08-08","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10407743/pdf/","citationCount":"2","resultStr":"{\"title\":\"Predicting molecular mechanisms of hereditary diseases by using their tissue-selective manifestation.\",\"authors\":\"Eyal Simonovsky, Moran Sharon, Maya Ziv, Omry Mauer, Idan Hekselman, Juman Jubran, Ekaterina Vinogradov, Chanan M Argov, Omer Basha, Lior Kerber, Yuval Yogev, Ayellet V Segrè, Hae Kyung Im, Ohad Birk, Lior Rokach, Esti Yeger-Lotem\",\"doi\":\"10.15252/msb.202211407\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>How do aberrations in widely expressed genes lead to tissue-selective hereditary diseases? Previous attempts to answer this question were limited to testing a few candidate mechanisms. To answer this question at a larger scale, we developed \\\"Tissue Risk Assessment of Causality by Expression\\\" (TRACE), a machine learning approach to predict genes that underlie tissue-selective diseases and selectivity-related features. TRACE utilized 4,744 biologically interpretable tissue-specific gene features that were inferred from heterogeneous omics datasets. Application of TRACE to 1,031 disease genes uncovered known and novel selectivity-related features, the most common of which was previously overlooked. Next, we created a catalog of tissue-associated risks for 18,927 protein-coding genes (https://netbio.bgu.ac.il/trace/). As proof-of-concept, we prioritized candidate disease genes identified in 48 rare-disease patients. TRACE ranked the verified disease gene among the patient's candidate genes significantly better than gene prioritization methods that rank by gene constraint or tissue expression. Thus, tissue selectivity combined with machine learning enhances genetic and clinical understanding of hereditary diseases.</p>\",\"PeriodicalId\":18906,\"journal\":{\"name\":\"Molecular Systems Biology\",\"volume\":\"19 8\",\"pages\":\"e11407\"},\"PeriodicalIF\":7.7000,\"publicationDate\":\"2023-08-08\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10407743/pdf/\",\"citationCount\":\"2\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Molecular Systems Biology\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://doi.org/10.15252/msb.202211407\",\"RegionNum\":1,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2023/5/26 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"Q1\",\"JCRName\":\"BIOCHEMISTRY & MOLECULAR BIOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Molecular Systems Biology","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.15252/msb.202211407","RegionNum":1,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2023/5/26 0:00:00","PubModel":"Epub","JCR":"Q1","JCRName":"BIOCHEMISTRY & MOLECULAR BIOLOGY","Score":null,"Total":0}
Predicting molecular mechanisms of hereditary diseases by using their tissue-selective manifestation.
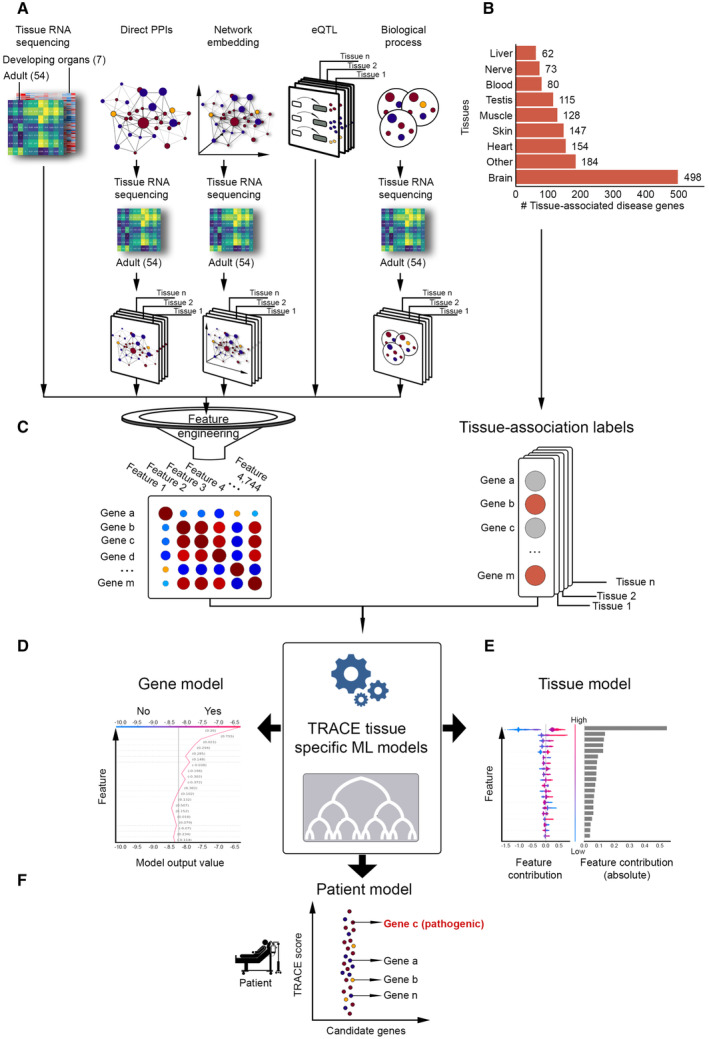
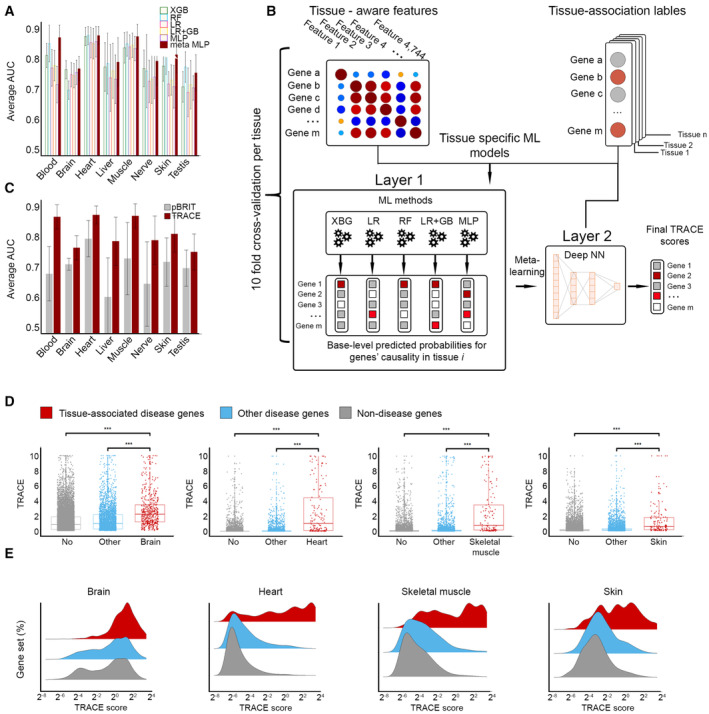
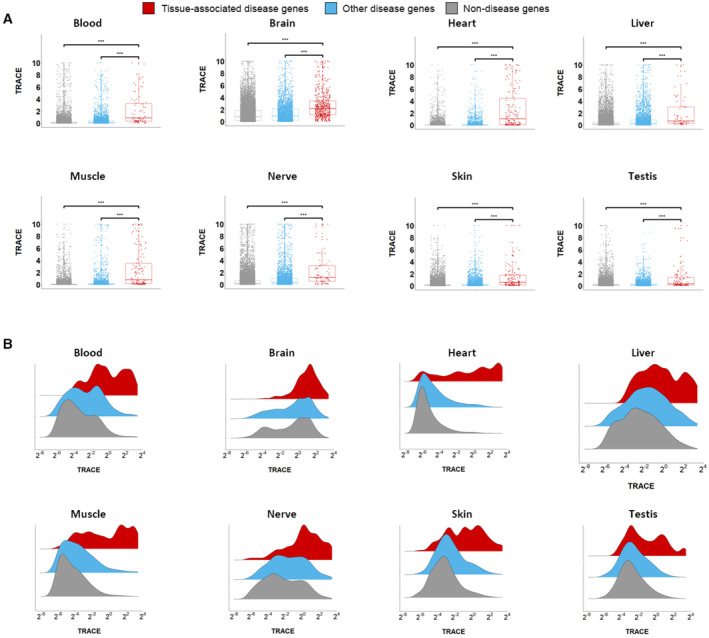
How do aberrations in widely expressed genes lead to tissue-selective hereditary diseases? Previous attempts to answer this question were limited to testing a few candidate mechanisms. To answer this question at a larger scale, we developed "Tissue Risk Assessment of Causality by Expression" (TRACE), a machine learning approach to predict genes that underlie tissue-selective diseases and selectivity-related features. TRACE utilized 4,744 biologically interpretable tissue-specific gene features that were inferred from heterogeneous omics datasets. Application of TRACE to 1,031 disease genes uncovered known and novel selectivity-related features, the most common of which was previously overlooked. Next, we created a catalog of tissue-associated risks for 18,927 protein-coding genes (https://netbio.bgu.ac.il/trace/). As proof-of-concept, we prioritized candidate disease genes identified in 48 rare-disease patients. TRACE ranked the verified disease gene among the patient's candidate genes significantly better than gene prioritization methods that rank by gene constraint or tissue expression. Thus, tissue selectivity combined with machine learning enhances genetic and clinical understanding of hereditary diseases.
期刊介绍:
Systems biology is a field that aims to understand complex biological systems by studying their components and how they interact. It is an integrative discipline that seeks to explain the properties and behavior of these systems.
Molecular Systems Biology is a scholarly journal that publishes top-notch research in the areas of systems biology, synthetic biology, and systems medicine. It is an open access journal, meaning that its content is freely available to readers, and it is peer-reviewed to ensure the quality of the published work.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
