{"title":"在蛋白质界面预测中优化选择合适的模板。","authors":"Steven Grudman, J Eduardo Fajardo, Andras Fiser","doi":"10.1093/bioinformatics/btad510","DOIUrl":null,"url":null,"abstract":"<p><strong>Motivation: </strong>Molecular-level classification of protein-protein interfaces can greatly assist in functional characterization and rational drug design. The most accurate protein interface predictions rely on finding homologous proteins with known interfaces since most interfaces are conserved within the same protein family. The accuracy of these template-based prediction approaches depends on the correct choice of suitable templates. Choosing the right templates in the immunoglobulin superfamily (IgSF) is challenging because its members share low sequence identity and display a wide range of alternative binding sites despite structural homology.</p><p><strong>Results: </strong>We present a new approach to predict protein interfaces. First, template-specific, informative evolutionary profiles are established using a mutual information-based approach. Next, based on the similarity of residue level conservation scores derived from the evolutionary profiles, a query protein is hierarchically clustered with all available template proteins in its superfamily with known interface definitions. Once clustered, a subset of the most closely related templates is selected, and an interface prediction is made. These initial interface predictions are subsequently refined by extensive docking. This method was benchmarked on 51 IgSF proteins and can predict nontrivial interfaces of IgSF proteins with an average and median F-score of 0.64 and 0.78, respectively. We also provide a way to assess the confidence of the results. The average and median F-scores increase to 0.8 and 0.81, respectively, if 27% of low confidence cases and 17% of medium confidence cases are removed. Lastly, we provide residue level interface predictions, protein complexes, and confidence measurements for singletons in the IgSF.</p><p><strong>Availability and implementation: </strong>Source code is freely available at: https://gitlab.com/fiserlab.org/interdct_with_refinement.</p>","PeriodicalId":8903,"journal":{"name":"Bioinformatics","volume":"39 9","pages":""},"PeriodicalIF":4.4000,"publicationDate":"2023-09-02","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10491951/pdf/","citationCount":"0","resultStr":"{\"title\":\"Optimal selection of suitable templates in protein interface prediction.\",\"authors\":\"Steven Grudman, J Eduardo Fajardo, Andras Fiser\",\"doi\":\"10.1093/bioinformatics/btad510\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Motivation: </strong>Molecular-level classification of protein-protein interfaces can greatly assist in functional characterization and rational drug design. The most accurate protein interface predictions rely on finding homologous proteins with known interfaces since most interfaces are conserved within the same protein family. The accuracy of these template-based prediction approaches depends on the correct choice of suitable templates. Choosing the right templates in the immunoglobulin superfamily (IgSF) is challenging because its members share low sequence identity and display a wide range of alternative binding sites despite structural homology.</p><p><strong>Results: </strong>We present a new approach to predict protein interfaces. First, template-specific, informative evolutionary profiles are established using a mutual information-based approach. Next, based on the similarity of residue level conservation scores derived from the evolutionary profiles, a query protein is hierarchically clustered with all available template proteins in its superfamily with known interface definitions. Once clustered, a subset of the most closely related templates is selected, and an interface prediction is made. These initial interface predictions are subsequently refined by extensive docking. This method was benchmarked on 51 IgSF proteins and can predict nontrivial interfaces of IgSF proteins with an average and median F-score of 0.64 and 0.78, respectively. We also provide a way to assess the confidence of the results. The average and median F-scores increase to 0.8 and 0.81, respectively, if 27% of low confidence cases and 17% of medium confidence cases are removed. Lastly, we provide residue level interface predictions, protein complexes, and confidence measurements for singletons in the IgSF.</p><p><strong>Availability and implementation: </strong>Source code is freely available at: https://gitlab.com/fiserlab.org/interdct_with_refinement.</p>\",\"PeriodicalId\":8903,\"journal\":{\"name\":\"Bioinformatics\",\"volume\":\"39 9\",\"pages\":\"\"},\"PeriodicalIF\":4.4000,\"publicationDate\":\"2023-09-02\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10491951/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Bioinformatics\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://doi.org/10.1093/bioinformatics/btad510\",\"RegionNum\":3,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"BIOCHEMICAL RESEARCH METHODS\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Bioinformatics","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1093/bioinformatics/btad510","RegionNum":3,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"BIOCHEMICAL RESEARCH METHODS","Score":null,"Total":0}
引用次数: 0
摘要
动机:蛋白质-蛋白质界面的分子级分类对功能表征和合理药物设计大有帮助。最准确的蛋白质界面预测依赖于寻找具有已知界面的同源蛋白质,因为大多数界面在同一蛋白质家族中是保守的。这些基于模板的预测方法的准确性取决于正确选择合适的模板。在免疫球蛋白超家族(IgSF)中选择合适的模板具有挑战性,因为其成员的序列同一性很低,而且尽管结构同源,但却显示出广泛的替代结合位点:结果:我们提出了一种预测蛋白质界面的新方法。结果:我们提出了预测蛋白质界面的新方法。首先,利用基于互信息的方法建立了特定模板的信息进化曲线。接下来,根据从进化图谱中得出的残基水平保护得分的相似性,将查询蛋白质与其超家族中已知界面定义的所有可用模板蛋白质进行分层聚类。聚类完成后,选择一个关系最密切的模板子集,并进行界面预测。随后通过广泛的对接来完善这些初步的界面预测。该方法在 51 个 IgSF 蛋白上进行了基准测试,可以预测 IgSF 蛋白的非复杂界面,平均 F 分数和中位数分别为 0.64 和 0.78。我们还提供了一种评估结果置信度的方法。如果去除 27% 的低置信度案例和 17% 的中等置信度案例,平均 F score 和中位 F score 分别增至 0.8 和 0.81。最后,我们还提供了残基级界面预测、蛋白质复合物以及 IgSF 中单体的置信度测量:源代码可在 https://gitlab.com/fiserlab.org/interdct_with_refinement 免费获取。
Optimal selection of suitable templates in protein interface prediction.
Motivation: Molecular-level classification of protein-protein interfaces can greatly assist in functional characterization and rational drug design. The most accurate protein interface predictions rely on finding homologous proteins with known interfaces since most interfaces are conserved within the same protein family. The accuracy of these template-based prediction approaches depends on the correct choice of suitable templates. Choosing the right templates in the immunoglobulin superfamily (IgSF) is challenging because its members share low sequence identity and display a wide range of alternative binding sites despite structural homology.
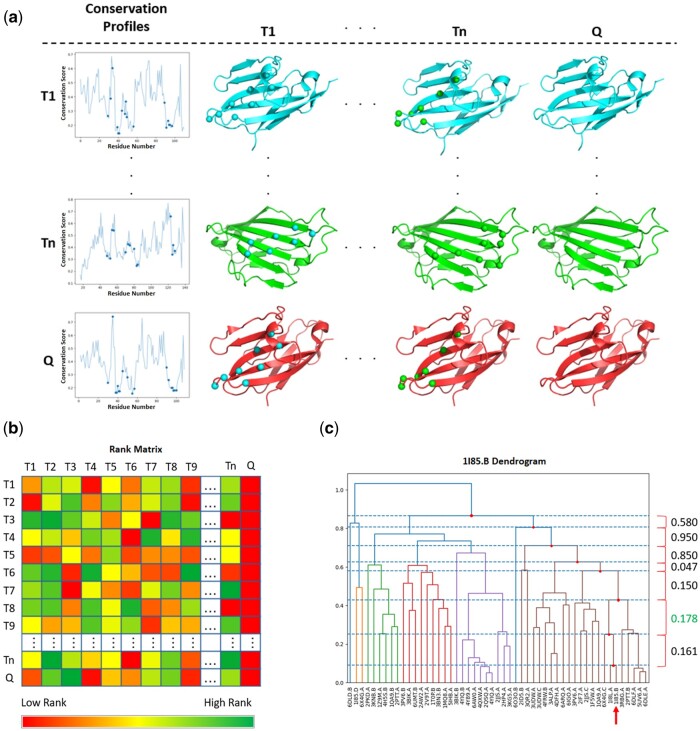
Results: We present a new approach to predict protein interfaces. First, template-specific, informative evolutionary profiles are established using a mutual information-based approach. Next, based on the similarity of residue level conservation scores derived from the evolutionary profiles, a query protein is hierarchically clustered with all available template proteins in its superfamily with known interface definitions. Once clustered, a subset of the most closely related templates is selected, and an interface prediction is made. These initial interface predictions are subsequently refined by extensive docking. This method was benchmarked on 51 IgSF proteins and can predict nontrivial interfaces of IgSF proteins with an average and median F-score of 0.64 and 0.78, respectively. We also provide a way to assess the confidence of the results. The average and median F-scores increase to 0.8 and 0.81, respectively, if 27% of low confidence cases and 17% of medium confidence cases are removed. Lastly, we provide residue level interface predictions, protein complexes, and confidence measurements for singletons in the IgSF.
Availability and implementation: Source code is freely available at: https://gitlab.com/fiserlab.org/interdct_with_refinement.
期刊介绍:
The leading journal in its field, Bioinformatics publishes the highest quality scientific papers and review articles of interest to academic and industrial researchers. Its main focus is on new developments in genome bioinformatics and computational biology. Two distinct sections within the journal - Discovery Notes and Application Notes- focus on shorter papers; the former reporting biologically interesting discoveries using computational methods, the latter exploring the applications used for experiments.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
