Jiangfen Wu, Lingyan Ren, Xinyi Huang, Li Hu, Liangliang Zhang, Dan Xie, Zhimin Li, Naijian Han, Shengwen Huang
{"title":"中国杜氏肌营养不良症家族DMD基因两个新变异的鉴定。","authors":"Jiangfen Wu, Lingyan Ren, Xinyi Huang, Li Hu, Liangliang Zhang, Dan Xie, Zhimin Li, Naijian Han, Shengwen Huang","doi":"10.2147/PGPM.S416294","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Duchenne muscular dystrophy (DMD), an X-linked recessive neuromuscular disorder, is caused by pathogenic variants in the <i>DMD</i> gene encoding a large structural protein in muscle cells.</p><p><strong>Methods: </strong>Two probands, a 6-year old boy and a 1-month old infant, respectively, were clinically diagnosed with DMD based on elevated levels of creatine kinase and creatine kinase isoenzyme. CNVplex and whole exome sequencing (WES) were performed for causal variants, and Sanger sequencing was used for verification.</p><p><strong>Results: </strong>CNVplex found no large deletions or duplications in the <i>DMD</i> gene in both patients, but WES discovered a single-nucleotide deletion in exon 48 (NM_004006.2:c.6963del, p.Asp2322ThrfsTer16) in the proband of pedigree 1, and a nonsense mutation in exon 27 (NM_004006.2:c.3637A>T, p.K1213Ter) in the proband of pedigree 2.</p><p><strong>Conclusion: </strong>The results of our study expand the mutation spectrum of DMD and enrich our understanding of the clinical characteristics of DMD. Genetic counseling was provided for the two families involved in this study.</p>","PeriodicalId":56015,"journal":{"name":"Pharmacogenomics & Personalized Medicine","volume":"16 ","pages":"759-766"},"PeriodicalIF":1.8000,"publicationDate":"2023-01-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/cc/c8/pgpm-16-759.PMC10441636.pdf","citationCount":"0","resultStr":"{\"title\":\"Identification of Two Novel Variants of the <i>DMD</i> Gene in Chinese Families with Duchenne Muscular Dystrophy.\",\"authors\":\"Jiangfen Wu, Lingyan Ren, Xinyi Huang, Li Hu, Liangliang Zhang, Dan Xie, Zhimin Li, Naijian Han, Shengwen Huang\",\"doi\":\"10.2147/PGPM.S416294\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Background: </strong>Duchenne muscular dystrophy (DMD), an X-linked recessive neuromuscular disorder, is caused by pathogenic variants in the <i>DMD</i> gene encoding a large structural protein in muscle cells.</p><p><strong>Methods: </strong>Two probands, a 6-year old boy and a 1-month old infant, respectively, were clinically diagnosed with DMD based on elevated levels of creatine kinase and creatine kinase isoenzyme. CNVplex and whole exome sequencing (WES) were performed for causal variants, and Sanger sequencing was used for verification.</p><p><strong>Results: </strong>CNVplex found no large deletions or duplications in the <i>DMD</i> gene in both patients, but WES discovered a single-nucleotide deletion in exon 48 (NM_004006.2:c.6963del, p.Asp2322ThrfsTer16) in the proband of pedigree 1, and a nonsense mutation in exon 27 (NM_004006.2:c.3637A>T, p.K1213Ter) in the proband of pedigree 2.</p><p><strong>Conclusion: </strong>The results of our study expand the mutation spectrum of DMD and enrich our understanding of the clinical characteristics of DMD. Genetic counseling was provided for the two families involved in this study.</p>\",\"PeriodicalId\":56015,\"journal\":{\"name\":\"Pharmacogenomics & Personalized Medicine\",\"volume\":\"16 \",\"pages\":\"759-766\"},\"PeriodicalIF\":1.8000,\"publicationDate\":\"2023-01-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/cc/c8/pgpm-16-759.PMC10441636.pdf\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Pharmacogenomics & Personalized Medicine\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://doi.org/10.2147/PGPM.S416294\",\"RegionNum\":4,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q3\",\"JCRName\":\"PHARMACOLOGY & PHARMACY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Pharmacogenomics & Personalized Medicine","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.2147/PGPM.S416294","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"PHARMACOLOGY & PHARMACY","Score":null,"Total":0}
Identification of Two Novel Variants of the DMD Gene in Chinese Families with Duchenne Muscular Dystrophy.
Background: Duchenne muscular dystrophy (DMD), an X-linked recessive neuromuscular disorder, is caused by pathogenic variants in the DMD gene encoding a large structural protein in muscle cells.
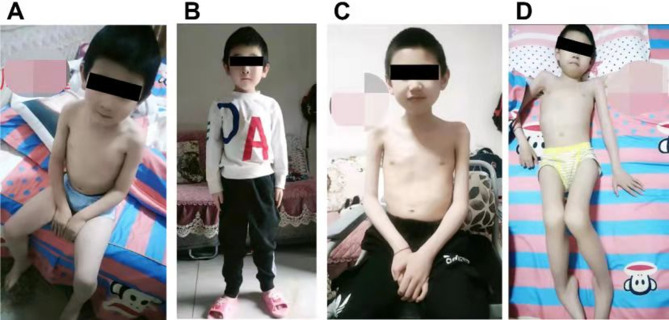
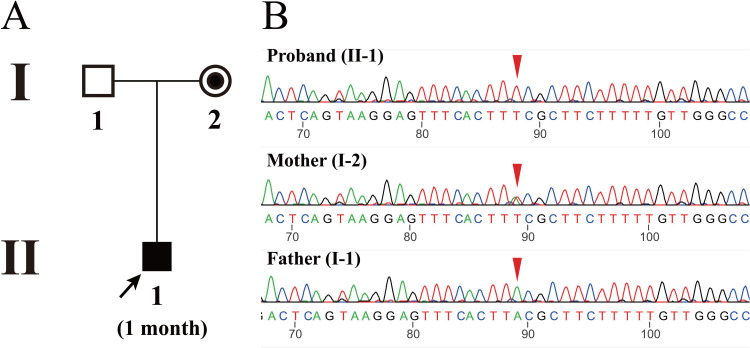
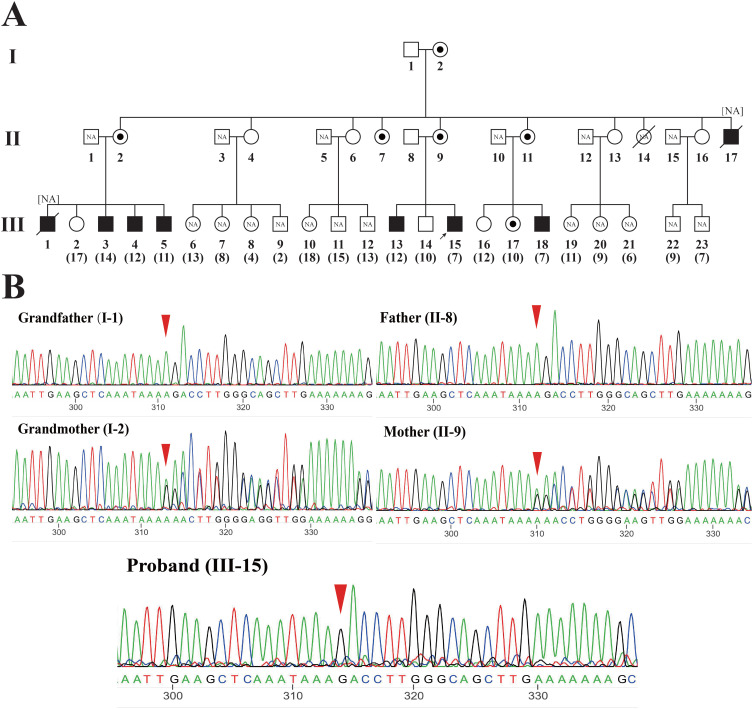
Methods: Two probands, a 6-year old boy and a 1-month old infant, respectively, were clinically diagnosed with DMD based on elevated levels of creatine kinase and creatine kinase isoenzyme. CNVplex and whole exome sequencing (WES) were performed for causal variants, and Sanger sequencing was used for verification.
Results: CNVplex found no large deletions or duplications in the DMD gene in both patients, but WES discovered a single-nucleotide deletion in exon 48 (NM_004006.2:c.6963del, p.Asp2322ThrfsTer16) in the proband of pedigree 1, and a nonsense mutation in exon 27 (NM_004006.2:c.3637A>T, p.K1213Ter) in the proband of pedigree 2.
Conclusion: The results of our study expand the mutation spectrum of DMD and enrich our understanding of the clinical characteristics of DMD. Genetic counseling was provided for the two families involved in this study.
期刊介绍:
Pharmacogenomics and Personalized Medicine is an international, peer-reviewed, open-access journal characterizing the influence of genotype on pharmacology leading to the development of personalized treatment programs and individualized drug selection for improved safety, efficacy and sustainability.
In particular, emphasis will be given to:
Genomic and proteomic profiling
Genetics and drug metabolism
Targeted drug identification and discovery
Optimizing drug selection & dosage based on patient''s genetic profile
Drug related morbidity & mortality intervention
Advanced disease screening and targeted therapeutic intervention
Genetic based vaccine development
Patient satisfaction and preference
Health economic evaluations
Practical and organizational issues in the development and implementation of personalized medicine programs.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
