{"title":"糖原储存病1a型动物模型研究综述","authors":"Irina O Petrova, Svetlana A Smirnikhina","doi":"10.1007/s11248-022-00325-7","DOIUrl":null,"url":null,"abstract":"<p><p>Glycogen storage disease type 1 (GSD1) is a rare hereditary monogenic disease characterized by the disturbed glucose metabolism. The most widespread variant of GSD1 is GSD1a, which is a deficiency of glucose-6-phosphatase-ɑ. Glucose-6-phosphatase-ɑ is expressed only in liver, kidney, and intestine, and these organs are primarily affected by its deficiency, and long-term complications of GSD1a include hepatic tumors and chronic liver disease. This article is a brief overview of existing animal models for GSD1a, from the first mouse model of 1996 to modern CRISPR/Cas9-generated ones. First whole-body murine models demonstrated exact metabolic symptoms of GSD1a, but the animals did not survive weaning. The protocol for glucose treatment allowed prolonged survival of affected animals, but long-term complications, such as hepatic tumorigenesis, could not be investigated. Next, organ-specific knockout models were developed, and most of the metabolic research was performed on liver glucose-6-phosphate-deficient mice. Naturally occuring mutation was also discovered in dogs. All these models are widely used to study GSD1a from metabolic and physiological standpoints and to develop possible treatments involving gene therapy. Research performed using these models helped elucidate the role of glycogen and lipid accumulation, hypoxia, mitochondrial dysfunction, and autophagy impairment in long-term complications of GSD1a, including hepatic tumorigenesis. Recently, gene replacement therapy and genome editing were tested on described models, and some of the developed approaches have reached clinical trials.</p>","PeriodicalId":23258,"journal":{"name":"Transgenic Research","volume":"31 6","pages":"593-606"},"PeriodicalIF":2.7000,"publicationDate":"2022-12-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"1","resultStr":"{\"title\":\"Studies on glycogen storage disease type 1a animal models: a brief perspective.\",\"authors\":\"Irina O Petrova, Svetlana A Smirnikhina\",\"doi\":\"10.1007/s11248-022-00325-7\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Glycogen storage disease type 1 (GSD1) is a rare hereditary monogenic disease characterized by the disturbed glucose metabolism. The most widespread variant of GSD1 is GSD1a, which is a deficiency of glucose-6-phosphatase-ɑ. Glucose-6-phosphatase-ɑ is expressed only in liver, kidney, and intestine, and these organs are primarily affected by its deficiency, and long-term complications of GSD1a include hepatic tumors and chronic liver disease. This article is a brief overview of existing animal models for GSD1a, from the first mouse model of 1996 to modern CRISPR/Cas9-generated ones. First whole-body murine models demonstrated exact metabolic symptoms of GSD1a, but the animals did not survive weaning. The protocol for glucose treatment allowed prolonged survival of affected animals, but long-term complications, such as hepatic tumorigenesis, could not be investigated. Next, organ-specific knockout models were developed, and most of the metabolic research was performed on liver glucose-6-phosphate-deficient mice. Naturally occuring mutation was also discovered in dogs. All these models are widely used to study GSD1a from metabolic and physiological standpoints and to develop possible treatments involving gene therapy. Research performed using these models helped elucidate the role of glycogen and lipid accumulation, hypoxia, mitochondrial dysfunction, and autophagy impairment in long-term complications of GSD1a, including hepatic tumorigenesis. Recently, gene replacement therapy and genome editing were tested on described models, and some of the developed approaches have reached clinical trials.</p>\",\"PeriodicalId\":23258,\"journal\":{\"name\":\"Transgenic Research\",\"volume\":\"31 6\",\"pages\":\"593-606\"},\"PeriodicalIF\":2.7000,\"publicationDate\":\"2022-12-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"1\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Transgenic Research\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://doi.org/10.1007/s11248-022-00325-7\",\"RegionNum\":3,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"BIOCHEMICAL RESEARCH METHODS\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Transgenic Research","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1007/s11248-022-00325-7","RegionNum":3,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"BIOCHEMICAL RESEARCH METHODS","Score":null,"Total":0}
Studies on glycogen storage disease type 1a animal models: a brief perspective.
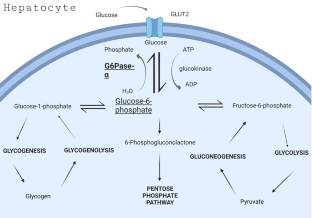
Glycogen storage disease type 1 (GSD1) is a rare hereditary monogenic disease characterized by the disturbed glucose metabolism. The most widespread variant of GSD1 is GSD1a, which is a deficiency of glucose-6-phosphatase-ɑ. Glucose-6-phosphatase-ɑ is expressed only in liver, kidney, and intestine, and these organs are primarily affected by its deficiency, and long-term complications of GSD1a include hepatic tumors and chronic liver disease. This article is a brief overview of existing animal models for GSD1a, from the first mouse model of 1996 to modern CRISPR/Cas9-generated ones. First whole-body murine models demonstrated exact metabolic symptoms of GSD1a, but the animals did not survive weaning. The protocol for glucose treatment allowed prolonged survival of affected animals, but long-term complications, such as hepatic tumorigenesis, could not be investigated. Next, organ-specific knockout models were developed, and most of the metabolic research was performed on liver glucose-6-phosphate-deficient mice. Naturally occuring mutation was also discovered in dogs. All these models are widely used to study GSD1a from metabolic and physiological standpoints and to develop possible treatments involving gene therapy. Research performed using these models helped elucidate the role of glycogen and lipid accumulation, hypoxia, mitochondrial dysfunction, and autophagy impairment in long-term complications of GSD1a, including hepatic tumorigenesis. Recently, gene replacement therapy and genome editing were tested on described models, and some of the developed approaches have reached clinical trials.
期刊介绍:
Transgenic Research focusses on transgenic and genome edited higher organisms. Manuscripts emphasizing biotechnological applications are strongly encouraged. Intellectual property, ethical issues, societal impact and regulatory aspects also fall within the scope of the journal. Transgenic Research aims to bridge the gap between fundamental and applied science in molecular biology and biotechnology for the plant and animal academic and associated industry communities.
Transgenic Research publishes
-Original Papers
-Reviews:
Should critically summarize the current state-of-the-art of the subject in a dispassionate way. Authors are requested to contact a Board Member before submission. Reviews should not be descriptive; rather they should present the most up-to-date information on the subject in a dispassionate and critical way. Perspective Reviews which can address new or controversial aspects are encouraged.
-Brief Communications:
Should report significant developments in methodology and experimental transgenic higher organisms



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
