Hiroya Naruse, So Okubo, Atsushi Sudo, Jun Mitsui, Takashi Mikata, Hiroyuki Ishiura, Shinichi Morishita, Shoji Tsuji, Tatsushi Toda
{"title":"携带HSPB1 p.Pro39Leu变异的迟发性远端遗传性运动神经病家族的临床特征","authors":"Hiroya Naruse, So Okubo, Atsushi Sudo, Jun Mitsui, Takashi Mikata, Hiroyuki Ishiura, Shinichi Morishita, Shoji Tsuji, Tatsushi Toda","doi":"10.1111/jns.12567","DOIUrl":null,"url":null,"abstract":"<div>\n \n \n <section>\n \n <h3> Background and Aims</h3>\n \n <p>Pathogenic variants of <i>HSPB1</i>, the gene encoding the small heat shock protein 27, have been reported to cause autosomal dominant distal hereditary motor neuropathy (dHMN) type II and autosomal dominant Charcot–Marie-Tooth (CMT) disease with minimal sensory involvement (CMT2F). This study aimed to describe the clinical features of patients in a family with late-onset dHMN carrying the Pro39Leu variant of <i>HSPB1</i>.</p>\n </section>\n \n <section>\n \n <h3> Methods</h3>\n \n <p>Whole-exome sequence analysis identified a heterozygous pathogenic variant (Pro39Leu) of <i>HSPB1</i> in the proband. The presence of the <i>HSPB1</i> Pro39Leu variant in two affected individuals was confirmed using direct nucleotide sequence analysis.</p>\n </section>\n \n <section>\n \n <h3> Results</h3>\n \n <p>Both patients exhibited distal muscle weakness with lower extremity predominance and no obvious sensory deficits, leading to a clinical diagnosis of late-onset dHMN. Nerve conduction studies (NCSs) revealed a subclinical complication of sensory disturbance in one of the patients. The clinical and electrophysiological findings of patients with the <i>HSPB1</i> Pro39Leu variant in this study and previous reports are summarized.</p>\n </section>\n \n <section>\n \n <h3> Interpretation</h3>\n \n <p>This study suggests that the clinical spectrum of patients carrying <i>HSPB1</i> Pro39Leu variants, especially the disease onset, might be broader than expected, and <i>HSPB1</i> variants should be considered in patients diagnosed with late-onset dHMN. Furthermore, patients with dHMN may have concomitant sensory deficits that should be evaluated using NCSs.</p>\n </section>\n </div>","PeriodicalId":17451,"journal":{"name":"Journal of the Peripheral Nervous System","volume":"28 3","pages":"518-521"},"PeriodicalIF":3.2000,"publicationDate":"2023-05-30","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1111/jns.12567","citationCount":"0","resultStr":"{\"title\":\"Clinical features of a family with late-onset distal hereditary motor neuropathy harboring p.Pro39Leu variant of HSPB1\",\"authors\":\"Hiroya Naruse, So Okubo, Atsushi Sudo, Jun Mitsui, Takashi Mikata, Hiroyuki Ishiura, Shinichi Morishita, Shoji Tsuji, Tatsushi Toda\",\"doi\":\"10.1111/jns.12567\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<div>\\n \\n \\n <section>\\n \\n <h3> Background and Aims</h3>\\n \\n <p>Pathogenic variants of <i>HSPB1</i>, the gene encoding the small heat shock protein 27, have been reported to cause autosomal dominant distal hereditary motor neuropathy (dHMN) type II and autosomal dominant Charcot–Marie-Tooth (CMT) disease with minimal sensory involvement (CMT2F). This study aimed to describe the clinical features of patients in a family with late-onset dHMN carrying the Pro39Leu variant of <i>HSPB1</i>.</p>\\n </section>\\n \\n <section>\\n \\n <h3> Methods</h3>\\n \\n <p>Whole-exome sequence analysis identified a heterozygous pathogenic variant (Pro39Leu) of <i>HSPB1</i> in the proband. The presence of the <i>HSPB1</i> Pro39Leu variant in two affected individuals was confirmed using direct nucleotide sequence analysis.</p>\\n </section>\\n \\n <section>\\n \\n <h3> Results</h3>\\n \\n <p>Both patients exhibited distal muscle weakness with lower extremity predominance and no obvious sensory deficits, leading to a clinical diagnosis of late-onset dHMN. Nerve conduction studies (NCSs) revealed a subclinical complication of sensory disturbance in one of the patients. The clinical and electrophysiological findings of patients with the <i>HSPB1</i> Pro39Leu variant in this study and previous reports are summarized.</p>\\n </section>\\n \\n <section>\\n \\n <h3> Interpretation</h3>\\n \\n <p>This study suggests that the clinical spectrum of patients carrying <i>HSPB1</i> Pro39Leu variants, especially the disease onset, might be broader than expected, and <i>HSPB1</i> variants should be considered in patients diagnosed with late-onset dHMN. Furthermore, patients with dHMN may have concomitant sensory deficits that should be evaluated using NCSs.</p>\\n </section>\\n </div>\",\"PeriodicalId\":17451,\"journal\":{\"name\":\"Journal of the Peripheral Nervous System\",\"volume\":\"28 3\",\"pages\":\"518-521\"},\"PeriodicalIF\":3.2000,\"publicationDate\":\"2023-05-30\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://onlinelibrary.wiley.com/doi/epdf/10.1111/jns.12567\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of the Peripheral Nervous System\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://onlinelibrary.wiley.com/doi/10.1111/jns.12567\",\"RegionNum\":3,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"CLINICAL NEUROLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of the Peripheral Nervous System","FirstCategoryId":"3","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1111/jns.12567","RegionNum":3,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CLINICAL NEUROLOGY","Score":null,"Total":0}
Clinical features of a family with late-onset distal hereditary motor neuropathy harboring p.Pro39Leu variant of HSPB1
Background and Aims
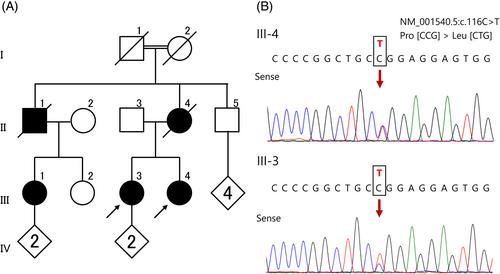
Pathogenic variants of HSPB1, the gene encoding the small heat shock protein 27, have been reported to cause autosomal dominant distal hereditary motor neuropathy (dHMN) type II and autosomal dominant Charcot–Marie-Tooth (CMT) disease with minimal sensory involvement (CMT2F). This study aimed to describe the clinical features of patients in a family with late-onset dHMN carrying the Pro39Leu variant of HSPB1.
Methods
Whole-exome sequence analysis identified a heterozygous pathogenic variant (Pro39Leu) of HSPB1 in the proband. The presence of the HSPB1 Pro39Leu variant in two affected individuals was confirmed using direct nucleotide sequence analysis.
Results
Both patients exhibited distal muscle weakness with lower extremity predominance and no obvious sensory deficits, leading to a clinical diagnosis of late-onset dHMN. Nerve conduction studies (NCSs) revealed a subclinical complication of sensory disturbance in one of the patients. The clinical and electrophysiological findings of patients with the HSPB1 Pro39Leu variant in this study and previous reports are summarized.
Interpretation
This study suggests that the clinical spectrum of patients carrying HSPB1 Pro39Leu variants, especially the disease onset, might be broader than expected, and HSPB1 variants should be considered in patients diagnosed with late-onset dHMN. Furthermore, patients with dHMN may have concomitant sensory deficits that should be evaluated using NCSs.
期刊介绍:
The Journal of the Peripheral Nervous System is the official journal of the Peripheral Nerve Society. Founded in 1996, it is the scientific journal of choice for clinicians, clinical scientists and basic neuroscientists interested in all aspects of biology and clinical research of peripheral nervous system disorders.
The Journal of the Peripheral Nervous System is a peer-reviewed journal that publishes high quality articles on cell and molecular biology, genomics, neuropathic pain, clinical research, trials, and unique case reports on inherited and acquired peripheral neuropathies.
Original articles are organized according to the topic in one of four specific areas: Mechanisms of Disease, Genetics, Clinical Research, and Clinical Trials.
The journal also publishes regular review papers on hot topics and Special Issues on basic, clinical, or assembled research in the field of peripheral nervous system disorders. Authors interested in contributing a review-type article or a Special Issue should contact the Editorial Office to discuss the scope of the proposed article with the Editor-in-Chief.


 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
