{"title":"ATN1标准hx基序外的新生变异个体的先天性张力低下和发育迟缓1例。","authors":"Elizaveta Makarova, Nicole R Legro, Ermal Aliu","doi":"10.1155/2023/1581876","DOIUrl":null,"url":null,"abstract":"<p><p>We present a case of a 4-year-old female with a de novo heterozygous variant in the ATN1 gene. The whole exome sequencing was performed on the patient and her parents, and a likely pathogenic, de novo variant was identified in exon 5 of the ATN1 gene. There are two well-documented conditions associated with the ATN1 gene: congenital hypotonia, epilepsy, developmental delay, and digital anomalies (CHEDDA) syndrome and dentatorubral-pallidoluysian atrophy (DRPLA). Unlike DRPLA which is caused by an expanded trinucleotide repeat, CHEDDA syndrome is caused by variants in the histidine-rich (HX) motif at exon 7 of ATN1 similar to the de novo variant found in exon 5 of the presented individual. CHEDDA syndrome is a neurodevelopmental disorder previously documented in over 17 unrelated individuals. Compared to other documented CHEDDA syndrome cases, this individual shares similarities in respect to hypotonia, hearing impairment, impaired gross and fine motor ability, gastrointestinal abnormalities, hyperextensible joints, and frontal bossing. However, the individual presented here has only a moderate developmental delay and has acquired more developmental milestones such as higher-level language skills and more developed fine motor skills, than previously described individuals. The authors of this paper believe the patient's milder phenotype may be due to the variant's location outside of the canonic HX motif.</p>","PeriodicalId":30325,"journal":{"name":"Case Reports in Genetics","volume":"2023 ","pages":"1581876"},"PeriodicalIF":0.0000,"publicationDate":"2023-01-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9845047/pdf/","citationCount":"0","resultStr":"{\"title\":\"A Case of Congenital Hypotonia and Developmental Delay in an Individual with a <i>De Novo</i> Variant Outside of the Canonical HX-Motif of ATN1.\",\"authors\":\"Elizaveta Makarova, Nicole R Legro, Ermal Aliu\",\"doi\":\"10.1155/2023/1581876\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>We present a case of a 4-year-old female with a de novo heterozygous variant in the ATN1 gene. The whole exome sequencing was performed on the patient and her parents, and a likely pathogenic, de novo variant was identified in exon 5 of the ATN1 gene. There are two well-documented conditions associated with the ATN1 gene: congenital hypotonia, epilepsy, developmental delay, and digital anomalies (CHEDDA) syndrome and dentatorubral-pallidoluysian atrophy (DRPLA). Unlike DRPLA which is caused by an expanded trinucleotide repeat, CHEDDA syndrome is caused by variants in the histidine-rich (HX) motif at exon 7 of ATN1 similar to the de novo variant found in exon 5 of the presented individual. CHEDDA syndrome is a neurodevelopmental disorder previously documented in over 17 unrelated individuals. Compared to other documented CHEDDA syndrome cases, this individual shares similarities in respect to hypotonia, hearing impairment, impaired gross and fine motor ability, gastrointestinal abnormalities, hyperextensible joints, and frontal bossing. However, the individual presented here has only a moderate developmental delay and has acquired more developmental milestones such as higher-level language skills and more developed fine motor skills, than previously described individuals. The authors of this paper believe the patient's milder phenotype may be due to the variant's location outside of the canonic HX motif.</p>\",\"PeriodicalId\":30325,\"journal\":{\"name\":\"Case Reports in Genetics\",\"volume\":\"2023 \",\"pages\":\"1581876\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2023-01-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9845047/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Case Reports in Genetics\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1155/2023/1581876\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"\",\"JCRName\":\"\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Case Reports in Genetics","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1155/2023/1581876","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"","JCRName":"","Score":null,"Total":0}
A Case of Congenital Hypotonia and Developmental Delay in an Individual with a De Novo Variant Outside of the Canonical HX-Motif of ATN1.
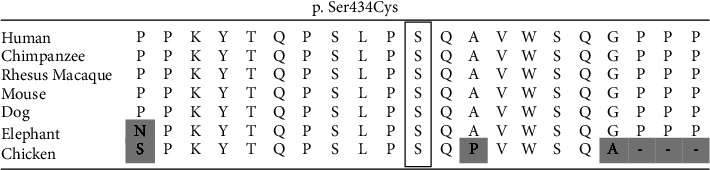
We present a case of a 4-year-old female with a de novo heterozygous variant in the ATN1 gene. The whole exome sequencing was performed on the patient and her parents, and a likely pathogenic, de novo variant was identified in exon 5 of the ATN1 gene. There are two well-documented conditions associated with the ATN1 gene: congenital hypotonia, epilepsy, developmental delay, and digital anomalies (CHEDDA) syndrome and dentatorubral-pallidoluysian atrophy (DRPLA). Unlike DRPLA which is caused by an expanded trinucleotide repeat, CHEDDA syndrome is caused by variants in the histidine-rich (HX) motif at exon 7 of ATN1 similar to the de novo variant found in exon 5 of the presented individual. CHEDDA syndrome is a neurodevelopmental disorder previously documented in over 17 unrelated individuals. Compared to other documented CHEDDA syndrome cases, this individual shares similarities in respect to hypotonia, hearing impairment, impaired gross and fine motor ability, gastrointestinal abnormalities, hyperextensible joints, and frontal bossing. However, the individual presented here has only a moderate developmental delay and has acquired more developmental milestones such as higher-level language skills and more developed fine motor skills, than previously described individuals. The authors of this paper believe the patient's milder phenotype may be due to the variant's location outside of the canonic HX motif.



 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
