Stephanie A Mercurio, Lauren M Chunn, Gus Khursigara, Catherine Nester, Kathleen Wray, Ulrike Botschen, Mark J Kiel, Frank Rutsch, Carlos R Ferreira
{"title":"ENPP1缺乏症:使用基因座特异性患者数据库的个体变异相关性的临床更新。","authors":"Stephanie A Mercurio, Lauren M Chunn, Gus Khursigara, Catherine Nester, Kathleen Wray, Ulrike Botschen, Mark J Kiel, Frank Rutsch, Carlos R Ferreira","doi":"10.1002/humu.24477","DOIUrl":null,"url":null,"abstract":"<p><p>Loss-of-function variants in the ectonucleotide pyrophosphatase/phosphodiesterase family member 1 (ENPP1) cause ENPP1 Deficiency, a rare disorder characterized by pathological calcification, neointimal proliferation, and impaired bone mineralization. The consequence of ENPP1 Deficiency is a broad range of age dependent symptoms and morbidities including cardiovascular complications and 50% mortality in infants, autosomal recessive hypophosphatemic rickets type 2 (ARHR2) in children, and joint pain, osteomalacia and enthesopathies in adults. Recent research continues to add to the growing clinical presentation profile as well as expanding the role of ENPP1 itself. Here we review the current knowledge on the spectrum of clinical and genetic findings of ENPP1 Deficiency reported in patients diagnosed with GACI or ARHR2 phenotypes using a comprehensive database of known ENPP1 variants with associated clinical data. A total of 108 genotypes were identified from 154 patients. Of the 109 ENPP1 variants reviewed, 72.5% were demonstrably disease-causing, a threefold increase in pathogenic/likely pathogenic variants over other databases. There is substantial heterogeneity in disease severity, even among patients with the same variant. The approach to creating a continuously curated database of ENPP1 variants accessible to clinicians is necessary to increase the diagnostic yield of clinical genetic testing and accelerate diagnosis of ENPP1 Deficiency.</p>","PeriodicalId":13061,"journal":{"name":"Human Mutation","volume":"43 12","pages":"1673-1705"},"PeriodicalIF":3.7000,"publicationDate":"2022-12-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12465099/pdf/","citationCount":"0","resultStr":"{\"title\":\"ENPP1 deficiency: A clinical update on the relevance of individual variants using a locus-specific patient database.\",\"authors\":\"Stephanie A Mercurio, Lauren M Chunn, Gus Khursigara, Catherine Nester, Kathleen Wray, Ulrike Botschen, Mark J Kiel, Frank Rutsch, Carlos R Ferreira\",\"doi\":\"10.1002/humu.24477\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Loss-of-function variants in the ectonucleotide pyrophosphatase/phosphodiesterase family member 1 (ENPP1) cause ENPP1 Deficiency, a rare disorder characterized by pathological calcification, neointimal proliferation, and impaired bone mineralization. The consequence of ENPP1 Deficiency is a broad range of age dependent symptoms and morbidities including cardiovascular complications and 50% mortality in infants, autosomal recessive hypophosphatemic rickets type 2 (ARHR2) in children, and joint pain, osteomalacia and enthesopathies in adults. Recent research continues to add to the growing clinical presentation profile as well as expanding the role of ENPP1 itself. Here we review the current knowledge on the spectrum of clinical and genetic findings of ENPP1 Deficiency reported in patients diagnosed with GACI or ARHR2 phenotypes using a comprehensive database of known ENPP1 variants with associated clinical data. A total of 108 genotypes were identified from 154 patients. Of the 109 ENPP1 variants reviewed, 72.5% were demonstrably disease-causing, a threefold increase in pathogenic/likely pathogenic variants over other databases. There is substantial heterogeneity in disease severity, even among patients with the same variant. The approach to creating a continuously curated database of ENPP1 variants accessible to clinicians is necessary to increase the diagnostic yield of clinical genetic testing and accelerate diagnosis of ENPP1 Deficiency.</p>\",\"PeriodicalId\":13061,\"journal\":{\"name\":\"Human Mutation\",\"volume\":\"43 12\",\"pages\":\"1673-1705\"},\"PeriodicalIF\":3.7000,\"publicationDate\":\"2022-12-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12465099/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Human Mutation\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://doi.org/10.1002/humu.24477\",\"RegionNum\":2,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2022/10/8 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"Q2\",\"JCRName\":\"GENETICS & HEREDITY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Human Mutation","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1002/humu.24477","RegionNum":2,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2022/10/8 0:00:00","PubModel":"Epub","JCR":"Q2","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
ENPP1 deficiency: A clinical update on the relevance of individual variants using a locus-specific patient database.
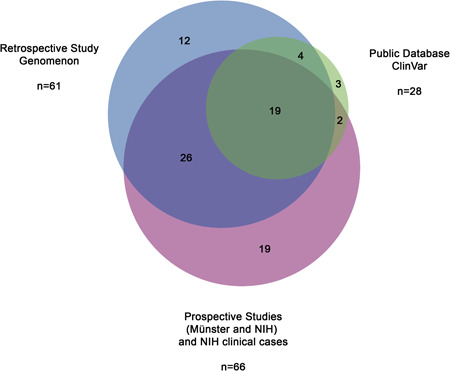
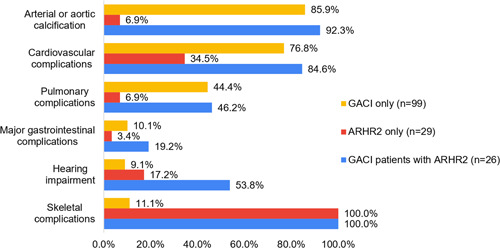
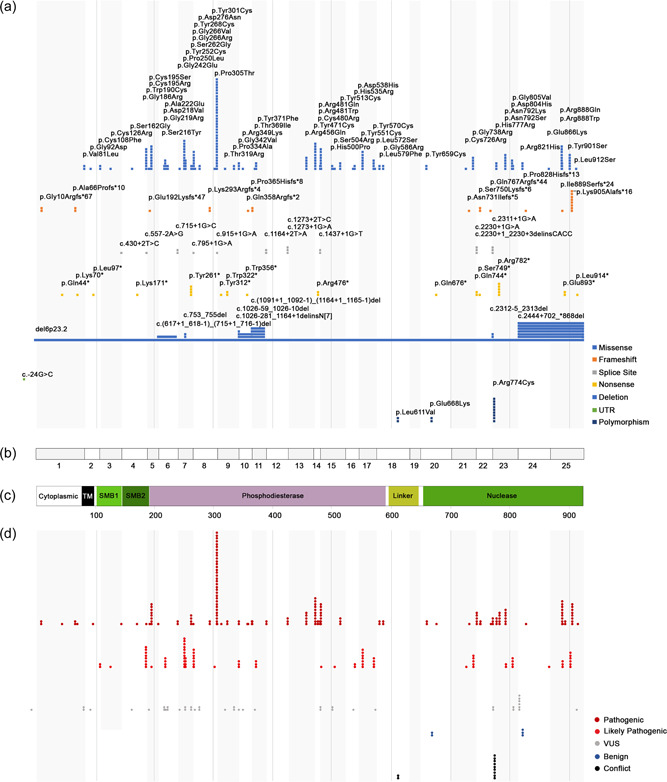
Loss-of-function variants in the ectonucleotide pyrophosphatase/phosphodiesterase family member 1 (ENPP1) cause ENPP1 Deficiency, a rare disorder characterized by pathological calcification, neointimal proliferation, and impaired bone mineralization. The consequence of ENPP1 Deficiency is a broad range of age dependent symptoms and morbidities including cardiovascular complications and 50% mortality in infants, autosomal recessive hypophosphatemic rickets type 2 (ARHR2) in children, and joint pain, osteomalacia and enthesopathies in adults. Recent research continues to add to the growing clinical presentation profile as well as expanding the role of ENPP1 itself. Here we review the current knowledge on the spectrum of clinical and genetic findings of ENPP1 Deficiency reported in patients diagnosed with GACI or ARHR2 phenotypes using a comprehensive database of known ENPP1 variants with associated clinical data. A total of 108 genotypes were identified from 154 patients. Of the 109 ENPP1 variants reviewed, 72.5% were demonstrably disease-causing, a threefold increase in pathogenic/likely pathogenic variants over other databases. There is substantial heterogeneity in disease severity, even among patients with the same variant. The approach to creating a continuously curated database of ENPP1 variants accessible to clinicians is necessary to increase the diagnostic yield of clinical genetic testing and accelerate diagnosis of ENPP1 Deficiency.
期刊介绍:
Human Mutation is a peer-reviewed journal that offers publication of original Research Articles, Methods, Mutation Updates, Reviews, Database Articles, Rapid Communications, and Letters on broad aspects of mutation research in humans. Reports of novel DNA variations and their phenotypic consequences, reports of SNPs demonstrated as valuable for genomic analysis, descriptions of new molecular detection methods, and novel approaches to clinical diagnosis are welcomed. Novel reports of gene organization at the genomic level, reported in the context of mutation investigation, may be considered. The journal provides a unique forum for the exchange of ideas, methods, and applications of interest to molecular, human, and medical geneticists in academic, industrial, and clinical research settings worldwide.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
