Nikolinka Yordanova, Violeta Iotova, Deborah J G Mackay, I Karen Temple, Sara Stoyanova, Mari Hachmeriyan
{"title":"一名晚期诊断的坦普尔综合征患者的长期随访--病例报告。","authors":"Nikolinka Yordanova, Violeta Iotova, Deborah J G Mackay, I Karen Temple, Sara Stoyanova, Mari Hachmeriyan","doi":"10.4274/jcrpe.galenos.2022.2022-9-19","DOIUrl":null,"url":null,"abstract":"<p><p>Temple syndrome is a rare imprinting disorder, caused by alterations in the critical imprinted region 14q32 of chromosome 14. It is characterized by pre- and postnatal growth retardation, truncal hypotonia and facial dysmorphism in the neonatal period. We report an 18-year-old girl with a late diagnosis of Temple syndrome presenting with all typical signs and symptoms including small for gestational age at birth, feeding difficulties, muscle hypotonia and delayed developmental milestones, central precocious puberty, truncal obesity and reduced growth. The patient is the second reported in the literature with signs of clinical and biochemical hyperandrogenism and the first treated with Dehydrocortisone<sup>®</sup>, with a good response. The clinical diagnosis of this patient was made after long-term follow up at a single center for rare endocrine diseases, and a molecular genetics diagnosis of complete hypomethylation of 14q32 chromosome imprinting center (DLK/GTL2) was recently established. Growth hormone treatment was not given and although precocious puberty was treated in line with standard protocols, her final height remained below the target range. Increased awareness of Temple syndrome and timely molecular diagnosis enables improvement of clinical care of these patients as well as prevention of inherent metabolic consequences.</p>","PeriodicalId":48805,"journal":{"name":"Journal of Clinical Research in Pediatric Endocrinology","volume":" ","pages":"475-480"},"PeriodicalIF":1.5000,"publicationDate":"2024-12-04","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11629717/pdf/","citationCount":"0","resultStr":"{\"title\":\"Long-term Follow-up of a Late Diagnosed Patient with Temple Syndrome\",\"authors\":\"Nikolinka Yordanova, Violeta Iotova, Deborah J G Mackay, I Karen Temple, Sara Stoyanova, Mari Hachmeriyan\",\"doi\":\"10.4274/jcrpe.galenos.2022.2022-9-19\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Temple syndrome is a rare imprinting disorder, caused by alterations in the critical imprinted region 14q32 of chromosome 14. It is characterized by pre- and postnatal growth retardation, truncal hypotonia and facial dysmorphism in the neonatal period. We report an 18-year-old girl with a late diagnosis of Temple syndrome presenting with all typical signs and symptoms including small for gestational age at birth, feeding difficulties, muscle hypotonia and delayed developmental milestones, central precocious puberty, truncal obesity and reduced growth. The patient is the second reported in the literature with signs of clinical and biochemical hyperandrogenism and the first treated with Dehydrocortisone<sup>®</sup>, with a good response. The clinical diagnosis of this patient was made after long-term follow up at a single center for rare endocrine diseases, and a molecular genetics diagnosis of complete hypomethylation of 14q32 chromosome imprinting center (DLK/GTL2) was recently established. Growth hormone treatment was not given and although precocious puberty was treated in line with standard protocols, her final height remained below the target range. Increased awareness of Temple syndrome and timely molecular diagnosis enables improvement of clinical care of these patients as well as prevention of inherent metabolic consequences.</p>\",\"PeriodicalId\":48805,\"journal\":{\"name\":\"Journal of Clinical Research in Pediatric Endocrinology\",\"volume\":\" \",\"pages\":\"475-480\"},\"PeriodicalIF\":1.5000,\"publicationDate\":\"2024-12-04\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11629717/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Clinical Research in Pediatric Endocrinology\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://doi.org/10.4274/jcrpe.galenos.2022.2022-9-19\",\"RegionNum\":4,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2023/2/2 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"Q4\",\"JCRName\":\"ENDOCRINOLOGY & METABOLISM\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Clinical Research in Pediatric Endocrinology","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.4274/jcrpe.galenos.2022.2022-9-19","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2023/2/2 0:00:00","PubModel":"Epub","JCR":"Q4","JCRName":"ENDOCRINOLOGY & METABOLISM","Score":null,"Total":0}
Long-term Follow-up of a Late Diagnosed Patient with Temple Syndrome
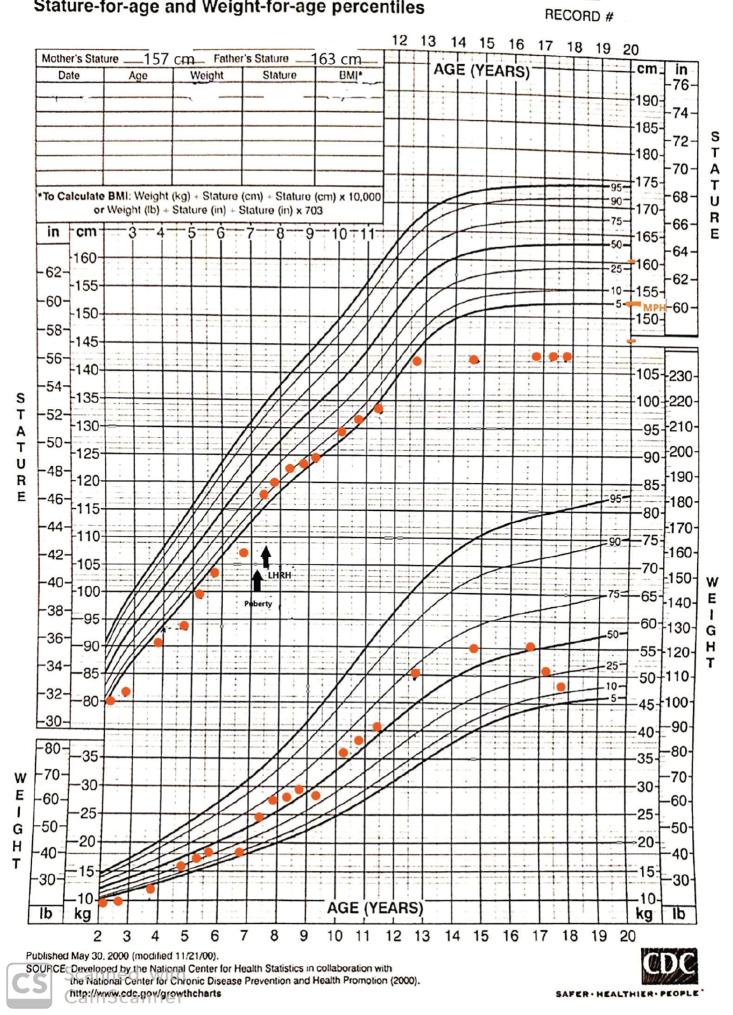
Temple syndrome is a rare imprinting disorder, caused by alterations in the critical imprinted region 14q32 of chromosome 14. It is characterized by pre- and postnatal growth retardation, truncal hypotonia and facial dysmorphism in the neonatal period. We report an 18-year-old girl with a late diagnosis of Temple syndrome presenting with all typical signs and symptoms including small for gestational age at birth, feeding difficulties, muscle hypotonia and delayed developmental milestones, central precocious puberty, truncal obesity and reduced growth. The patient is the second reported in the literature with signs of clinical and biochemical hyperandrogenism and the first treated with Dehydrocortisone®, with a good response. The clinical diagnosis of this patient was made after long-term follow up at a single center for rare endocrine diseases, and a molecular genetics diagnosis of complete hypomethylation of 14q32 chromosome imprinting center (DLK/GTL2) was recently established. Growth hormone treatment was not given and although precocious puberty was treated in line with standard protocols, her final height remained below the target range. Increased awareness of Temple syndrome and timely molecular diagnosis enables improvement of clinical care of these patients as well as prevention of inherent metabolic consequences.
期刊介绍:
The Journal of Clinical Research in Pediatric Endocrinology (JCRPE) publishes original research articles, reviews, short communications, letters, case reports and other special features related to the field of pediatric endocrinology. JCRPE is published in English by the Turkish Pediatric Endocrinology and Diabetes Society quarterly (March, June, September, December). The target audience is physicians, researchers and other healthcare professionals in all areas of pediatric endocrinology.





 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
