Luís Leite de Sousa, Gonçalo Pimenta, Rita Veríssimo, Tiago J Carvalho, Ivo Laranjinha
{"title":"邓氏病:肾衰竭的一种罕见病因。","authors":"Luís Leite de Sousa, Gonçalo Pimenta, Rita Veríssimo, Tiago J Carvalho, Ivo Laranjinha","doi":"10.5414/CNCS110975","DOIUrl":null,"url":null,"abstract":"<p><p>Dent's disease is an X-linked recessive disease characterized by proximal tubulopathy with low-molecular weight proteinuria, hypercalciuria, nephrolithiasis, nephrocalcinosis, and kidney failure. It is mainly caused by mutations in the <i>CLCN5</i> or <i>OCRL1</i> genes, and only ~ 250 families have been identified with these mutations. We present a 31-year-old male referred to a nephrology consultation due to elevated serum creatinine and a history of nephrolithiasis. Complementary evaluation revealed protein/creatinine ratio of 1.9 g/g and albumin/creatinine ratio of 0.5 g/g, hypercalciuria and medullary nephrocalcinosis. These findings raised the suspicion of Dent's disease, which was confirmed by genetic testing. A missense mutation in the <i>CLCN5</i> gene (c.810C>G, p.(Ser270Arg)), not previously reported in populational databases, was identified. During the evaluation of the patient, it came to our attention that a first-degree male cousin was being followed in our kidney transplantation unit. Given the unknown etiology of his chronic kidney disease, genetic testing was performed, identifying the same mutation. This case highlights the importance of considering the diagnosis of Dent's disease in the setting of a male patient with chronic kidney disease of unknown etiology, low-molecular-weight proteinuria, hypercalciuria, and nephrocalcinosis. Despite progression to end-stage kidney failure in a significant portion of male patients, there are no reports of recurrence after kidney transplantation.</p>","PeriodicalId":10398,"journal":{"name":"Clinical Nephrology. Case Studies","volume":"11 ","pages":"1-5"},"PeriodicalIF":0.0000,"publicationDate":"2023-01-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9850247/pdf/","citationCount":"0","resultStr":"{\"title\":\"Dent's disease: An unusual cause of kidney failure.\",\"authors\":\"Luís Leite de Sousa, Gonçalo Pimenta, Rita Veríssimo, Tiago J Carvalho, Ivo Laranjinha\",\"doi\":\"10.5414/CNCS110975\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Dent's disease is an X-linked recessive disease characterized by proximal tubulopathy with low-molecular weight proteinuria, hypercalciuria, nephrolithiasis, nephrocalcinosis, and kidney failure. It is mainly caused by mutations in the <i>CLCN5</i> or <i>OCRL1</i> genes, and only ~ 250 families have been identified with these mutations. We present a 31-year-old male referred to a nephrology consultation due to elevated serum creatinine and a history of nephrolithiasis. Complementary evaluation revealed protein/creatinine ratio of 1.9 g/g and albumin/creatinine ratio of 0.5 g/g, hypercalciuria and medullary nephrocalcinosis. These findings raised the suspicion of Dent's disease, which was confirmed by genetic testing. A missense mutation in the <i>CLCN5</i> gene (c.810C>G, p.(Ser270Arg)), not previously reported in populational databases, was identified. During the evaluation of the patient, it came to our attention that a first-degree male cousin was being followed in our kidney transplantation unit. Given the unknown etiology of his chronic kidney disease, genetic testing was performed, identifying the same mutation. This case highlights the importance of considering the diagnosis of Dent's disease in the setting of a male patient with chronic kidney disease of unknown etiology, low-molecular-weight proteinuria, hypercalciuria, and nephrocalcinosis. Despite progression to end-stage kidney failure in a significant portion of male patients, there are no reports of recurrence after kidney transplantation.</p>\",\"PeriodicalId\":10398,\"journal\":{\"name\":\"Clinical Nephrology. Case Studies\",\"volume\":\"11 \",\"pages\":\"1-5\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2023-01-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9850247/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Clinical Nephrology. Case Studies\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.5414/CNCS110975\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"\",\"JCRName\":\"\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Clinical Nephrology. Case Studies","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.5414/CNCS110975","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"","JCRName":"","Score":null,"Total":0}
Dent's disease: An unusual cause of kidney failure.
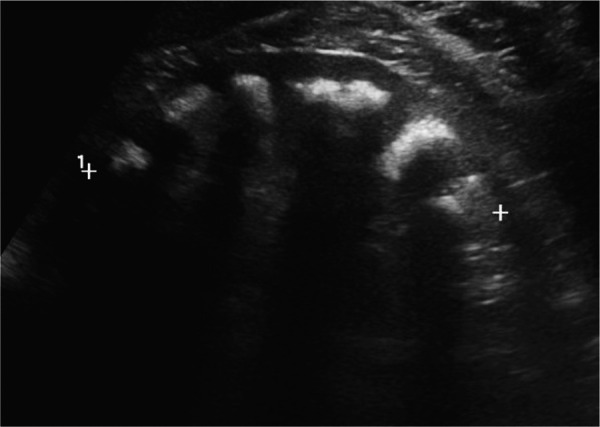
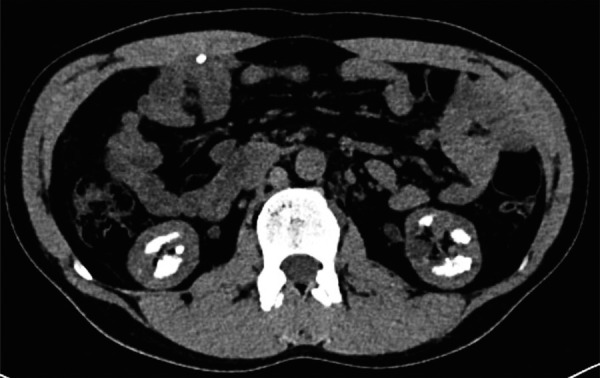
Dent's disease is an X-linked recessive disease characterized by proximal tubulopathy with low-molecular weight proteinuria, hypercalciuria, nephrolithiasis, nephrocalcinosis, and kidney failure. It is mainly caused by mutations in the CLCN5 or OCRL1 genes, and only ~ 250 families have been identified with these mutations. We present a 31-year-old male referred to a nephrology consultation due to elevated serum creatinine and a history of nephrolithiasis. Complementary evaluation revealed protein/creatinine ratio of 1.9 g/g and albumin/creatinine ratio of 0.5 g/g, hypercalciuria and medullary nephrocalcinosis. These findings raised the suspicion of Dent's disease, which was confirmed by genetic testing. A missense mutation in the CLCN5 gene (c.810C>G, p.(Ser270Arg)), not previously reported in populational databases, was identified. During the evaluation of the patient, it came to our attention that a first-degree male cousin was being followed in our kidney transplantation unit. Given the unknown etiology of his chronic kidney disease, genetic testing was performed, identifying the same mutation. This case highlights the importance of considering the diagnosis of Dent's disease in the setting of a male patient with chronic kidney disease of unknown etiology, low-molecular-weight proteinuria, hypercalciuria, and nephrocalcinosis. Despite progression to end-stage kidney failure in a significant portion of male patients, there are no reports of recurrence after kidney transplantation.




 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
