{"title":"1型多发性内分泌肿瘤的新疗法?","authors":"Hessa Boharoon, Ashley Grossman","doi":"10.17925/EE.2022.18.2.86","DOIUrl":null,"url":null,"abstract":"<p><p>Pancreatic neuroendocrine tumours (pNETs) are a major manifestation of multiple endocrine neoplasia type 1 (MEN1), and the most significant cause of morbidity and mortality in this disorder. There is some evidence that the early use of somatostatin analogues can retard progression, especially of small non-functioning tumours, but there are no other prophylactic therapies for patients, and the treatment of metastatic disease is similar to that for sporadic pNETs. A recent study has shown that in cell line and animal models, <i>MEN1</i> mutations lead to an upregulation of the enzyme dihydroorotate dehydrogenase (DHODH), which is involved in increasing precursor metabolites for the synthesis of pyrimidines. In these studies, blockade of this pathway by various means, including the DHODH inhibitor leflunomide, attenuates cell growth and tumour progression, suggesting a critical dependence on DHODH specifically in <i>MEN1</i>-mutated tissue. Preliminary clinical studies in three patients with MEN1 and pNETs have indicated some therapeutic potential of this drug, which has previously been used for some years in patients with rheumatoid arthritis. It is suggested that further clinical trials of this re-purposed drug are indicated to evaluate its potential for the treatment of patients with MEN1 and pNETS. This article describes the clinical problem of MEN1 and pNETs, and reviews the recent publication reporting on these initial results.</p>","PeriodicalId":75231,"journal":{"name":"TouchREVIEWS in endocrinology","volume":"18 2","pages":"86-88"},"PeriodicalIF":0.0000,"publicationDate":"2022-11-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9838189/pdf/","citationCount":"0","resultStr":"{\"title\":\"A New Medical Therapy for Multiple Endocrine Neoplasia Type 1?\",\"authors\":\"Hessa Boharoon, Ashley Grossman\",\"doi\":\"10.17925/EE.2022.18.2.86\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Pancreatic neuroendocrine tumours (pNETs) are a major manifestation of multiple endocrine neoplasia type 1 (MEN1), and the most significant cause of morbidity and mortality in this disorder. There is some evidence that the early use of somatostatin analogues can retard progression, especially of small non-functioning tumours, but there are no other prophylactic therapies for patients, and the treatment of metastatic disease is similar to that for sporadic pNETs. A recent study has shown that in cell line and animal models, <i>MEN1</i> mutations lead to an upregulation of the enzyme dihydroorotate dehydrogenase (DHODH), which is involved in increasing precursor metabolites for the synthesis of pyrimidines. In these studies, blockade of this pathway by various means, including the DHODH inhibitor leflunomide, attenuates cell growth and tumour progression, suggesting a critical dependence on DHODH specifically in <i>MEN1</i>-mutated tissue. Preliminary clinical studies in three patients with MEN1 and pNETs have indicated some therapeutic potential of this drug, which has previously been used for some years in patients with rheumatoid arthritis. It is suggested that further clinical trials of this re-purposed drug are indicated to evaluate its potential for the treatment of patients with MEN1 and pNETS. This article describes the clinical problem of MEN1 and pNETs, and reviews the recent publication reporting on these initial results.</p>\",\"PeriodicalId\":75231,\"journal\":{\"name\":\"TouchREVIEWS in endocrinology\",\"volume\":\"18 2\",\"pages\":\"86-88\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2022-11-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9838189/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"TouchREVIEWS in endocrinology\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.17925/EE.2022.18.2.86\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"\",\"JCRName\":\"\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"TouchREVIEWS in endocrinology","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.17925/EE.2022.18.2.86","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"","JCRName":"","Score":null,"Total":0}
A New Medical Therapy for Multiple Endocrine Neoplasia Type 1?
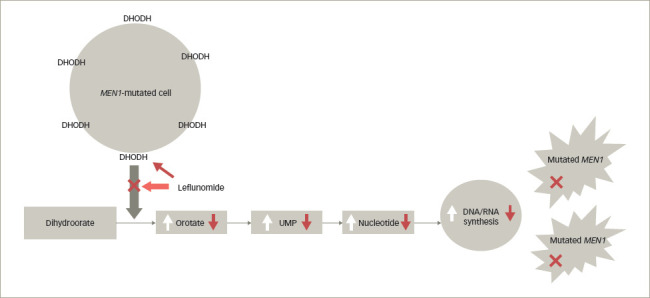
Pancreatic neuroendocrine tumours (pNETs) are a major manifestation of multiple endocrine neoplasia type 1 (MEN1), and the most significant cause of morbidity and mortality in this disorder. There is some evidence that the early use of somatostatin analogues can retard progression, especially of small non-functioning tumours, but there are no other prophylactic therapies for patients, and the treatment of metastatic disease is similar to that for sporadic pNETs. A recent study has shown that in cell line and animal models, MEN1 mutations lead to an upregulation of the enzyme dihydroorotate dehydrogenase (DHODH), which is involved in increasing precursor metabolites for the synthesis of pyrimidines. In these studies, blockade of this pathway by various means, including the DHODH inhibitor leflunomide, attenuates cell growth and tumour progression, suggesting a critical dependence on DHODH specifically in MEN1-mutated tissue. Preliminary clinical studies in three patients with MEN1 and pNETs have indicated some therapeutic potential of this drug, which has previously been used for some years in patients with rheumatoid arthritis. It is suggested that further clinical trials of this re-purposed drug are indicated to evaluate its potential for the treatment of patients with MEN1 and pNETS. This article describes the clinical problem of MEN1 and pNETs, and reviews the recent publication reporting on these initial results.



 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
