Matthew D Smith, Marshall A Case, Emily K Makowski, Peter M Tessier
{"title":"位置特异性富集比矩阵得分可从深度测序数据中预测抗体变异特性。","authors":"Matthew D Smith, Marshall A Case, Emily K Makowski, Peter M Tessier","doi":"10.1093/bioinformatics/btad446","DOIUrl":null,"url":null,"abstract":"<p><strong>Motivation: </strong>Deep sequencing of antibody and related protein libraries after phage or yeast-surface display sorting is widely used to identify variants with increased affinity, specificity, and/or improvements in key biophysical properties. Conventional approaches for identifying optimal variants typically use the frequencies of observation in enriched libraries or the corresponding enrichment ratios. However, these approaches disregard the vast majority of deep sequencing data and often fail to identify the best variants in the libraries.</p><p><strong>Results: </strong>Here, we present a method, Position-Specific Enrichment Ratio Matrix (PSERM) scoring, that uses entire deep sequencing datasets from pre- and post-selections to score each observed protein variant. The PSERM scores are the sum of the site-specific enrichment ratios observed at each mutated position. We find that PSERM scores are much more reproducible and correlate more strongly with experimentally measured properties than frequencies or enrichment ratios, including for multiple antibody properties (affinity and non-specific binding) for a clinical-stage antibody (emibetuzumab). We expect that this method will be broadly applicable to diverse protein engineering campaigns.</p><p><strong>Availability and implementation: </strong>All deep sequencing datasets and code to perform the analyses presented within are available via https://github.com/Tessier-Lab-UMich/PSERM_paper.</p>","PeriodicalId":8903,"journal":{"name":"Bioinformatics","volume":"39 9","pages":""},"PeriodicalIF":5.4000,"publicationDate":"2023-09-02","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10477941/pdf/","citationCount":"0","resultStr":"{\"title\":\"Position-Specific Enrichment Ratio Matrix scores predict antibody variant properties from deep sequencing data.\",\"authors\":\"Matthew D Smith, Marshall A Case, Emily K Makowski, Peter M Tessier\",\"doi\":\"10.1093/bioinformatics/btad446\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Motivation: </strong>Deep sequencing of antibody and related protein libraries after phage or yeast-surface display sorting is widely used to identify variants with increased affinity, specificity, and/or improvements in key biophysical properties. Conventional approaches for identifying optimal variants typically use the frequencies of observation in enriched libraries or the corresponding enrichment ratios. However, these approaches disregard the vast majority of deep sequencing data and often fail to identify the best variants in the libraries.</p><p><strong>Results: </strong>Here, we present a method, Position-Specific Enrichment Ratio Matrix (PSERM) scoring, that uses entire deep sequencing datasets from pre- and post-selections to score each observed protein variant. The PSERM scores are the sum of the site-specific enrichment ratios observed at each mutated position. We find that PSERM scores are much more reproducible and correlate more strongly with experimentally measured properties than frequencies or enrichment ratios, including for multiple antibody properties (affinity and non-specific binding) for a clinical-stage antibody (emibetuzumab). We expect that this method will be broadly applicable to diverse protein engineering campaigns.</p><p><strong>Availability and implementation: </strong>All deep sequencing datasets and code to perform the analyses presented within are available via https://github.com/Tessier-Lab-UMich/PSERM_paper.</p>\",\"PeriodicalId\":8903,\"journal\":{\"name\":\"Bioinformatics\",\"volume\":\"39 9\",\"pages\":\"\"},\"PeriodicalIF\":5.4000,\"publicationDate\":\"2023-09-02\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10477941/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Bioinformatics\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://doi.org/10.1093/bioinformatics/btad446\",\"RegionNum\":3,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"BIOCHEMICAL RESEARCH METHODS\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Bioinformatics","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1093/bioinformatics/btad446","RegionNum":3,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"BIOCHEMICAL RESEARCH METHODS","Score":null,"Total":0}
Position-Specific Enrichment Ratio Matrix scores predict antibody variant properties from deep sequencing data.
Motivation: Deep sequencing of antibody and related protein libraries after phage or yeast-surface display sorting is widely used to identify variants with increased affinity, specificity, and/or improvements in key biophysical properties. Conventional approaches for identifying optimal variants typically use the frequencies of observation in enriched libraries or the corresponding enrichment ratios. However, these approaches disregard the vast majority of deep sequencing data and often fail to identify the best variants in the libraries.
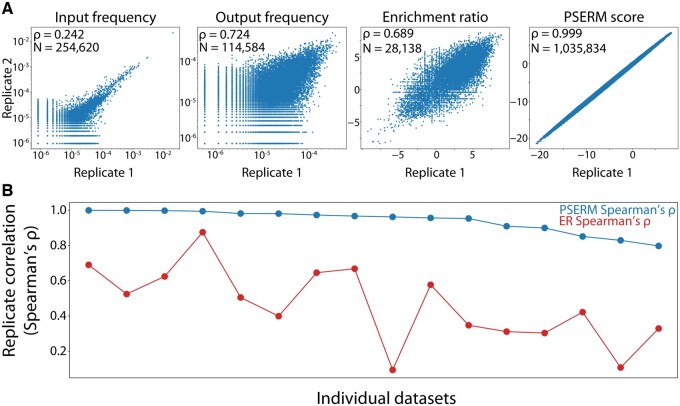
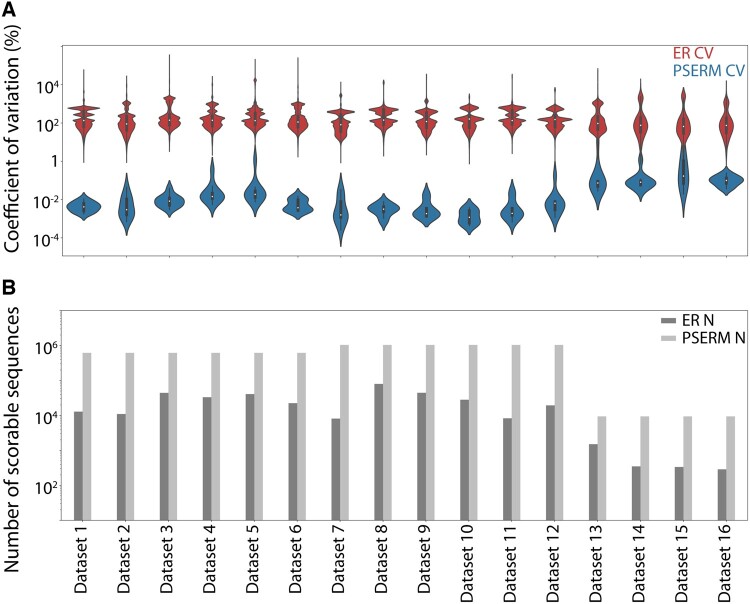
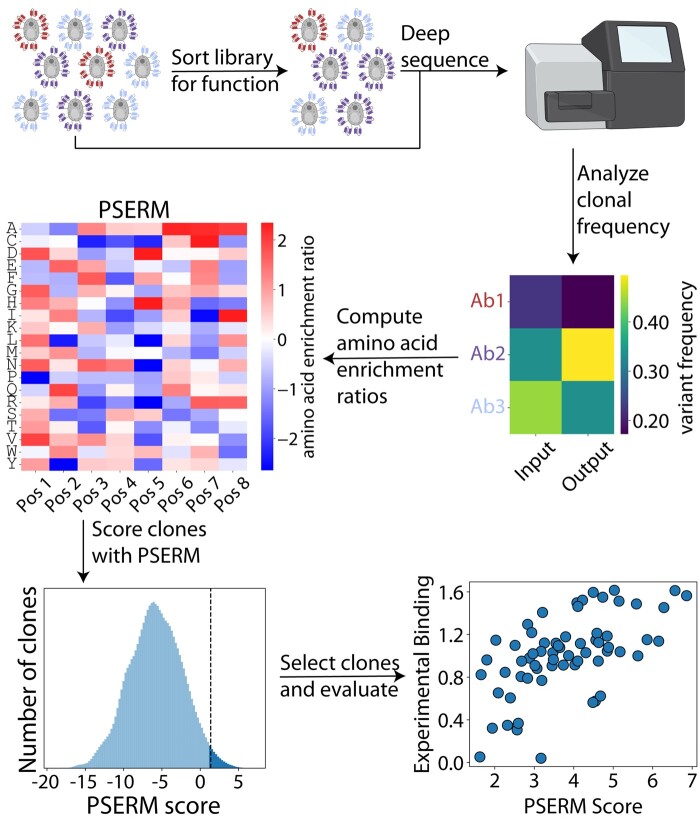
Results: Here, we present a method, Position-Specific Enrichment Ratio Matrix (PSERM) scoring, that uses entire deep sequencing datasets from pre- and post-selections to score each observed protein variant. The PSERM scores are the sum of the site-specific enrichment ratios observed at each mutated position. We find that PSERM scores are much more reproducible and correlate more strongly with experimentally measured properties than frequencies or enrichment ratios, including for multiple antibody properties (affinity and non-specific binding) for a clinical-stage antibody (emibetuzumab). We expect that this method will be broadly applicable to diverse protein engineering campaigns.
Availability and implementation: All deep sequencing datasets and code to perform the analyses presented within are available via https://github.com/Tessier-Lab-UMich/PSERM_paper.
期刊介绍:
The leading journal in its field, Bioinformatics publishes the highest quality scientific papers and review articles of interest to academic and industrial researchers. Its main focus is on new developments in genome bioinformatics and computational biology. Two distinct sections within the journal - Discovery Notes and Application Notes- focus on shorter papers; the former reporting biologically interesting discoveries using computational methods, the latter exploring the applications used for experiments.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
