{"title":"利用正则化回归建模路径间依赖关系的相干路径富集估计。","authors":"Kim Philipp Jablonski, Niko Beerenwinkel","doi":"10.1093/bioinformatics/btad522","DOIUrl":null,"url":null,"abstract":"<p><strong>Motivation: </strong>Gene set enrichment methods are a common tool to improve the interpretability of gene lists as obtained, for example, from differential gene expression analyses. They are based on computing whether dysregulated genes are located in certain biological pathways more often than expected by chance. Gene set enrichment tools rely on pre-existing pathway databases such as KEGG, Reactome, or the Gene Ontology. These databases are increasing in size and in the number of redundancies between pathways, which complicates the statistical enrichment computation.</p><p><strong>Results: </strong>We address this problem and develop a novel gene set enrichment method, called pareg, which is based on a regularized generalized linear model and directly incorporates dependencies between gene sets related to certain biological functions, for example, due to shared genes, in the enrichment computation. We show that pareg is more robust to noise than competing methods. Additionally, we demonstrate the ability of our method to recover known pathways as well as to suggest novel treatment targets in an exploratory analysis using breast cancer samples from TCGA.</p><p><strong>Availability and implementation: </strong>pareg is freely available as an R package on Bioconductor (https://bioconductor.org/packages/release/bioc/html/pareg.html) as well as on https://github.com/cbg-ethz/pareg. The GitHub repository also contains the Snakemake workflows needed to reproduce all results presented here.</p>","PeriodicalId":8903,"journal":{"name":"Bioinformatics","volume":"39 8","pages":""},"PeriodicalIF":5.4000,"publicationDate":"2023-08-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10471899/pdf/","citationCount":"0","resultStr":"{\"title\":\"Coherent pathway enrichment estimation by modeling inter-pathway dependencies using regularized regression.\",\"authors\":\"Kim Philipp Jablonski, Niko Beerenwinkel\",\"doi\":\"10.1093/bioinformatics/btad522\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Motivation: </strong>Gene set enrichment methods are a common tool to improve the interpretability of gene lists as obtained, for example, from differential gene expression analyses. They are based on computing whether dysregulated genes are located in certain biological pathways more often than expected by chance. Gene set enrichment tools rely on pre-existing pathway databases such as KEGG, Reactome, or the Gene Ontology. These databases are increasing in size and in the number of redundancies between pathways, which complicates the statistical enrichment computation.</p><p><strong>Results: </strong>We address this problem and develop a novel gene set enrichment method, called pareg, which is based on a regularized generalized linear model and directly incorporates dependencies between gene sets related to certain biological functions, for example, due to shared genes, in the enrichment computation. We show that pareg is more robust to noise than competing methods. Additionally, we demonstrate the ability of our method to recover known pathways as well as to suggest novel treatment targets in an exploratory analysis using breast cancer samples from TCGA.</p><p><strong>Availability and implementation: </strong>pareg is freely available as an R package on Bioconductor (https://bioconductor.org/packages/release/bioc/html/pareg.html) as well as on https://github.com/cbg-ethz/pareg. The GitHub repository also contains the Snakemake workflows needed to reproduce all results presented here.</p>\",\"PeriodicalId\":8903,\"journal\":{\"name\":\"Bioinformatics\",\"volume\":\"39 8\",\"pages\":\"\"},\"PeriodicalIF\":5.4000,\"publicationDate\":\"2023-08-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10471899/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Bioinformatics\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://doi.org/10.1093/bioinformatics/btad522\",\"RegionNum\":3,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"BIOCHEMICAL RESEARCH METHODS\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Bioinformatics","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1093/bioinformatics/btad522","RegionNum":3,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"BIOCHEMICAL RESEARCH METHODS","Score":null,"Total":0}
Coherent pathway enrichment estimation by modeling inter-pathway dependencies using regularized regression.
Motivation: Gene set enrichment methods are a common tool to improve the interpretability of gene lists as obtained, for example, from differential gene expression analyses. They are based on computing whether dysregulated genes are located in certain biological pathways more often than expected by chance. Gene set enrichment tools rely on pre-existing pathway databases such as KEGG, Reactome, or the Gene Ontology. These databases are increasing in size and in the number of redundancies between pathways, which complicates the statistical enrichment computation.
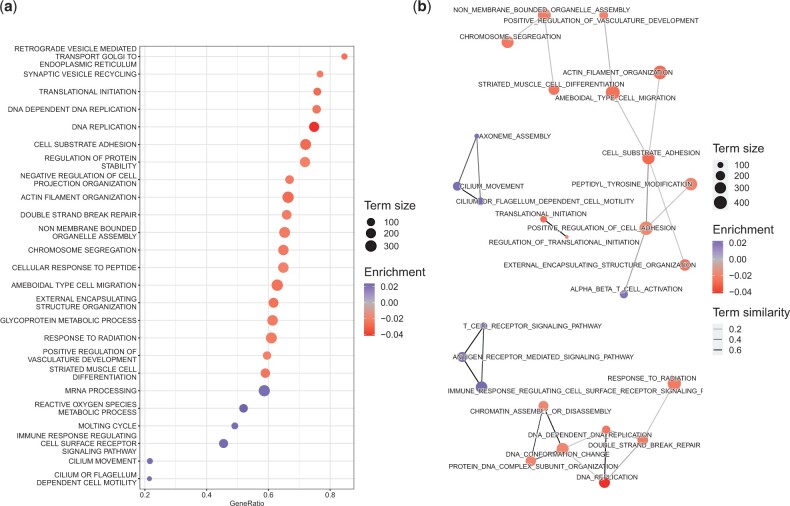
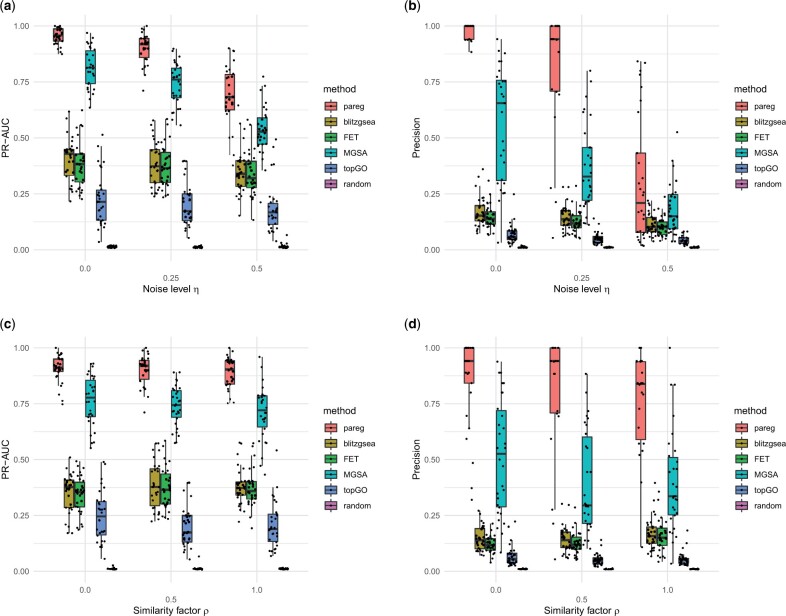
Results: We address this problem and develop a novel gene set enrichment method, called pareg, which is based on a regularized generalized linear model and directly incorporates dependencies between gene sets related to certain biological functions, for example, due to shared genes, in the enrichment computation. We show that pareg is more robust to noise than competing methods. Additionally, we demonstrate the ability of our method to recover known pathways as well as to suggest novel treatment targets in an exploratory analysis using breast cancer samples from TCGA.
Availability and implementation: pareg is freely available as an R package on Bioconductor (https://bioconductor.org/packages/release/bioc/html/pareg.html) as well as on https://github.com/cbg-ethz/pareg. The GitHub repository also contains the Snakemake workflows needed to reproduce all results presented here.
期刊介绍:
The leading journal in its field, Bioinformatics publishes the highest quality scientific papers and review articles of interest to academic and industrial researchers. Its main focus is on new developments in genome bioinformatics and computational biology. Two distinct sections within the journal - Discovery Notes and Application Notes- focus on shorter papers; the former reporting biologically interesting discoveries using computational methods, the latter exploring the applications used for experiments.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
