Maymunah Khries, Albert Lim, Dipayan Mitra, Mark Anderson, Jan Bengtsson, Ann Bowron, Elizabeth Harris, Jessica Blickwedel, Karen Wood, Anna P Basu
{"title":"SLC22A5表型谱的拓宽:原发性肉毒碱缺乏表现为局灶性肌阵挛。","authors":"Maymunah Khries, Albert Lim, Dipayan Mitra, Mark Anderson, Jan Bengtsson, Ann Bowron, Elizabeth Harris, Jessica Blickwedel, Karen Wood, Anna P Basu","doi":"10.1177/2329048X231184183","DOIUrl":null,"url":null,"abstract":"<p><p>Primary carnitine deficiency (PCD) is caused by pathogenic variants of the <i>SLC22A5</i> gene, which encodes a transmembrane protein that functions as a high affinity carnitine transporter. Carnitine is essential for the transport of acyl-CoA, produced from fatty acids, into the mitochondria where they are oxidised to produce energy. We present the case history of an 8-year-old boy who presented with fever, lethargy, focal rhythmic (3 Hz) left wrist twitching, and severe encephalopathy. MRI brain showed basal ganglia involvement. Metabolic investigations revealed low serum carnitine; whole genome sequencing confirmed compound heterozygous <i>SLC22A5</i> mutations. With carnitine replacement, intensive care support, and neurorehabilitation, he made a remarkable recovery, regaining independent breathing, speech, mobility, and hand use. Seizure presentation in PCD is rare and presentation with sustained focal myoclonus has not been previously reported. This case expands the known phenotype of PCD. Prompt carnitine replacement is imperative.</p>","PeriodicalId":72572,"journal":{"name":"Child neurology open","volume":"10 ","pages":"2329048X231184183"},"PeriodicalIF":0.0000,"publicationDate":"2023-01-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/bb/38/10.1177_2329048X231184183.PMC10354736.pdf","citationCount":"0","resultStr":"{\"title\":\"Broadening the Spectrum of <i>SLC22A5</i> Phenotype: Primary Carnitine Deficiency Presenting with Focal Myoclonus.\",\"authors\":\"Maymunah Khries, Albert Lim, Dipayan Mitra, Mark Anderson, Jan Bengtsson, Ann Bowron, Elizabeth Harris, Jessica Blickwedel, Karen Wood, Anna P Basu\",\"doi\":\"10.1177/2329048X231184183\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Primary carnitine deficiency (PCD) is caused by pathogenic variants of the <i>SLC22A5</i> gene, which encodes a transmembrane protein that functions as a high affinity carnitine transporter. Carnitine is essential for the transport of acyl-CoA, produced from fatty acids, into the mitochondria where they are oxidised to produce energy. We present the case history of an 8-year-old boy who presented with fever, lethargy, focal rhythmic (3 Hz) left wrist twitching, and severe encephalopathy. MRI brain showed basal ganglia involvement. Metabolic investigations revealed low serum carnitine; whole genome sequencing confirmed compound heterozygous <i>SLC22A5</i> mutations. With carnitine replacement, intensive care support, and neurorehabilitation, he made a remarkable recovery, regaining independent breathing, speech, mobility, and hand use. Seizure presentation in PCD is rare and presentation with sustained focal myoclonus has not been previously reported. This case expands the known phenotype of PCD. Prompt carnitine replacement is imperative.</p>\",\"PeriodicalId\":72572,\"journal\":{\"name\":\"Child neurology open\",\"volume\":\"10 \",\"pages\":\"2329048X231184183\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2023-01-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/bb/38/10.1177_2329048X231184183.PMC10354736.pdf\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Child neurology open\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1177/2329048X231184183\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"\",\"JCRName\":\"\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Child neurology open","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1177/2329048X231184183","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"","JCRName":"","Score":null,"Total":0}
Broadening the Spectrum of SLC22A5 Phenotype: Primary Carnitine Deficiency Presenting with Focal Myoclonus.
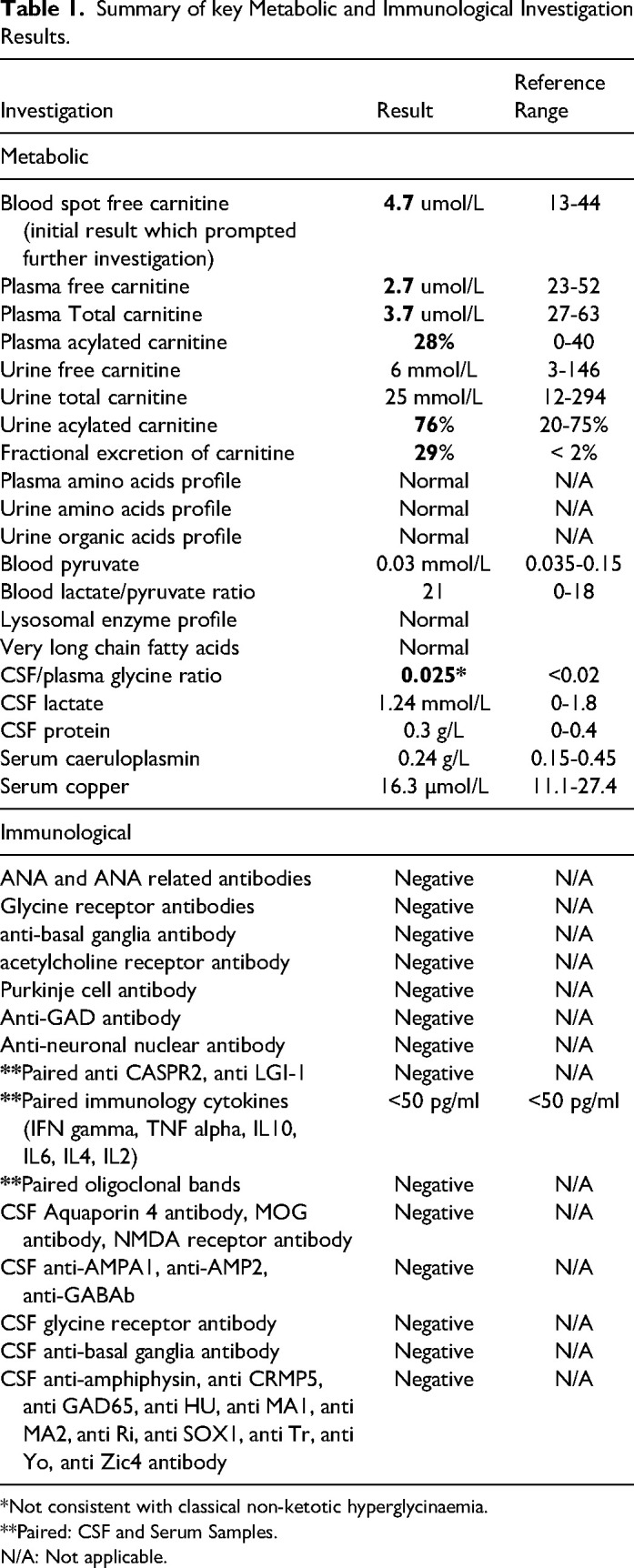
Primary carnitine deficiency (PCD) is caused by pathogenic variants of the SLC22A5 gene, which encodes a transmembrane protein that functions as a high affinity carnitine transporter. Carnitine is essential for the transport of acyl-CoA, produced from fatty acids, into the mitochondria where they are oxidised to produce energy. We present the case history of an 8-year-old boy who presented with fever, lethargy, focal rhythmic (3 Hz) left wrist twitching, and severe encephalopathy. MRI brain showed basal ganglia involvement. Metabolic investigations revealed low serum carnitine; whole genome sequencing confirmed compound heterozygous SLC22A5 mutations. With carnitine replacement, intensive care support, and neurorehabilitation, he made a remarkable recovery, regaining independent breathing, speech, mobility, and hand use. Seizure presentation in PCD is rare and presentation with sustained focal myoclonus has not been previously reported. This case expands the known phenotype of PCD. Prompt carnitine replacement is imperative.



 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
