Mythily Ganapathi, Christie M Buchovecky, Fernando Cristo, Priyanka Ahimaz, Carrie Ruzal-Shapiro, Karen Wou, José M Inácio, Alejandro Iglesias, José A Belo, Vaidehi Jobanputra
{"title":"一种新的双等位基因功能丧失变异导致DAND5异位综合征。","authors":"Mythily Ganapathi, Christie M Buchovecky, Fernando Cristo, Priyanka Ahimaz, Carrie Ruzal-Shapiro, Karen Wou, José M Inácio, Alejandro Iglesias, José A Belo, Vaidehi Jobanputra","doi":"10.1101/mcs.a006248","DOIUrl":null,"url":null,"abstract":"<p><p>The majority of heterotaxy cases do not obtain a molecular diagnosis, although pathogenic variants in more than 50 genes are known to cause heterotaxy. A heterozygous missense variant in <i>DAND5</i>, a nodal inhibitor, which functions in early development for establishment of right-left patterning, has been implicated in heterotaxy. Recently, the first case was reported of a <i>DAND5</i> biallelic loss-of-function (LoF) variant in an individual with heterotaxy. Here, we describe a second unrelated individual with heterotaxy syndrome and a homozygous frameshift variant in <i>DAND5</i> (NM_152654.2:c.197del [p.Leu66ArgfsTer22]). Using an in vitro assay, we demonstrate that the <i>DAND5</i> c.197del variant is unable to inhibit nodal signaling when compared with the wild-type expression construct. This work strengthens the genetic and functional evidence for biallelic LoF variants in <i>DAND5</i> causing an autosomal recessive heterotaxy syndrome.</p>","PeriodicalId":10360,"journal":{"name":"Cold Spring Harbor Molecular Case Studies","volume":"8 7","pages":""},"PeriodicalIF":1.8000,"publicationDate":"2022-12-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/13/6a/MCS006248Gan.PMC9808554.pdf","citationCount":"0","resultStr":"{\"title\":\"A novel biallelic loss-of-function variant in <i>DAND5</i> causes heterotaxy syndrome.\",\"authors\":\"Mythily Ganapathi, Christie M Buchovecky, Fernando Cristo, Priyanka Ahimaz, Carrie Ruzal-Shapiro, Karen Wou, José M Inácio, Alejandro Iglesias, José A Belo, Vaidehi Jobanputra\",\"doi\":\"10.1101/mcs.a006248\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>The majority of heterotaxy cases do not obtain a molecular diagnosis, although pathogenic variants in more than 50 genes are known to cause heterotaxy. A heterozygous missense variant in <i>DAND5</i>, a nodal inhibitor, which functions in early development for establishment of right-left patterning, has been implicated in heterotaxy. Recently, the first case was reported of a <i>DAND5</i> biallelic loss-of-function (LoF) variant in an individual with heterotaxy. Here, we describe a second unrelated individual with heterotaxy syndrome and a homozygous frameshift variant in <i>DAND5</i> (NM_152654.2:c.197del [p.Leu66ArgfsTer22]). Using an in vitro assay, we demonstrate that the <i>DAND5</i> c.197del variant is unable to inhibit nodal signaling when compared with the wild-type expression construct. This work strengthens the genetic and functional evidence for biallelic LoF variants in <i>DAND5</i> causing an autosomal recessive heterotaxy syndrome.</p>\",\"PeriodicalId\":10360,\"journal\":{\"name\":\"Cold Spring Harbor Molecular Case Studies\",\"volume\":\"8 7\",\"pages\":\"\"},\"PeriodicalIF\":1.8000,\"publicationDate\":\"2022-12-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/13/6a/MCS006248Gan.PMC9808554.pdf\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Cold Spring Harbor Molecular Case Studies\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1101/mcs.a006248\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q3\",\"JCRName\":\"MEDICINE, RESEARCH & EXPERIMENTAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Cold Spring Harbor Molecular Case Studies","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1101/mcs.a006248","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"MEDICINE, RESEARCH & EXPERIMENTAL","Score":null,"Total":0}
A novel biallelic loss-of-function variant in DAND5 causes heterotaxy syndrome.
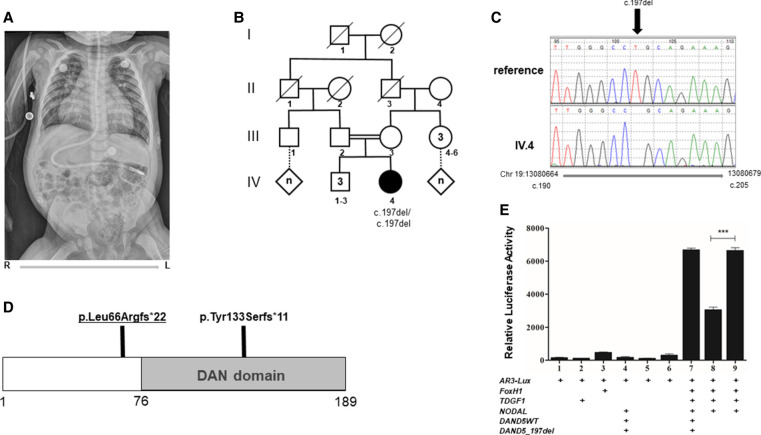
The majority of heterotaxy cases do not obtain a molecular diagnosis, although pathogenic variants in more than 50 genes are known to cause heterotaxy. A heterozygous missense variant in DAND5, a nodal inhibitor, which functions in early development for establishment of right-left patterning, has been implicated in heterotaxy. Recently, the first case was reported of a DAND5 biallelic loss-of-function (LoF) variant in an individual with heterotaxy. Here, we describe a second unrelated individual with heterotaxy syndrome and a homozygous frameshift variant in DAND5 (NM_152654.2:c.197del [p.Leu66ArgfsTer22]). Using an in vitro assay, we demonstrate that the DAND5 c.197del variant is unable to inhibit nodal signaling when compared with the wild-type expression construct. This work strengthens the genetic and functional evidence for biallelic LoF variants in DAND5 causing an autosomal recessive heterotaxy syndrome.
期刊介绍:
Cold Spring Harbor Molecular Case Studies is an open-access, peer-reviewed, international journal in the field of precision medicine. Articles in the journal present genomic and molecular analyses of individuals or cohorts alongside their clinical presentations and phenotypic information. The journal''s purpose is to rapidly share insights into disease development and treatment gained by application of genomics, proteomics, metabolomics, biomarker analysis, and other approaches. The journal covers the fields of cancer, complex diseases, monogenic disorders, neurological conditions, orphan diseases, infectious disease, gene therapy, and pharmacogenomics. It has a rapid peer-review process that is based on technical evaluation of the analyses performed, not the novelty of findings, and offers a swift, clear path to publication. The journal publishes: Research Reports presenting detailed case studies of individuals and small cohorts, Research Articles describing more extensive work using larger cohorts and/or functional analyses, Rapid Communications presenting the discovery of a novel variant and/or novel phenotype associated with a known disease gene, Rapid Cancer Communications presenting the discovery of a novel variant or combination of variants in a cancer type, Variant Discrepancy Resolution describing efforts to resolve differences or update variant interpretations in ClinVar through case-level data sharing, Follow-up Reports linked to previous observations, Plus Review Articles, Editorials, and Position Statements on best practices for research in precision medicine.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
