Ozan Çiftçi, Cornelis A M Wagemaker, Adrienne Mertens, Peter van Bodegom, Walter Pirovano, Barbara Gravendeel
{"title":"模拟群落中硅藻类群相对丰度的基因分型分析。","authors":"Ozan Çiftçi, Cornelis A M Wagemaker, Adrienne Mertens, Peter van Bodegom, Walter Pirovano, Barbara Gravendeel","doi":"10.1186/s12862-023-02104-2","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Diatoms are present in all waters and are highly sensitive to pollution gradients. Therefore, they are ideal bioindicators for water quality assessment. Current indices used in these applications are based on identifying diatom species and counting their abundances using traditional light microscopy. Several molecular techniques have been developed to help automate different steps of this process, but obtaining reliable estimates of diatom community composition and species abundance remains challenging.</p><p><strong>Results: </strong>Here, we evaluated a recently developed quantification method based on Genotyping by Sequencing (GBS) for the first time in diatoms to estimate the relative abundances within a species complex. For this purpose, a reference database comprised of thousands of genomic DNA clusters was generated from cultures of Nitzschia palea. The sequencing reads from calibration and mock samples were mapped against this database for parallel quantification. We sequenced 25 mock diatom communities containing up to five taxa per sample in different abundances. Taxon abundances in these communities were also quantified by a diatom expert using manual counting of cells on light microscopic slides. The relative abundances of strains across mock samples were over- or under-estimated by the manual counting method, and a majority of mock samples had stronger correlations using GBS. Moreover, one previously recognized putative hybrid had the largest number of false positive detections demonstrating the limitation of the manual counting method when morphologically similar and/or phylogenetically close taxa are analyzed.</p><p><strong>Conclusions: </strong>Our results suggest that GBS is a reliable method to estimate the relative abundances of the N. palea taxa analyzed in this study and outperformed traditional light microscopy in terms of accuracy. GBS provides increased taxonomic resolution compared to currently available quantitative molecular approaches, and it is more scalable in the number of species that can be analyzed in a single run. Hence, this is a significant step forward in developing automated, high-throughput molecular methods specifically designed for the quantification of [diatom] communities for freshwater quality assessments.</p>","PeriodicalId":9127,"journal":{"name":"BMC Ecology and Evolution","volume":"23 1","pages":"4"},"PeriodicalIF":0.0000,"publicationDate":"2023-02-06","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9903628/pdf/","citationCount":"0","resultStr":"{\"title\":\"Genotyping by sequencing for estimating relative abundances of diatom taxa in mock communities.\",\"authors\":\"Ozan Çiftçi, Cornelis A M Wagemaker, Adrienne Mertens, Peter van Bodegom, Walter Pirovano, Barbara Gravendeel\",\"doi\":\"10.1186/s12862-023-02104-2\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Background: </strong>Diatoms are present in all waters and are highly sensitive to pollution gradients. Therefore, they are ideal bioindicators for water quality assessment. Current indices used in these applications are based on identifying diatom species and counting their abundances using traditional light microscopy. Several molecular techniques have been developed to help automate different steps of this process, but obtaining reliable estimates of diatom community composition and species abundance remains challenging.</p><p><strong>Results: </strong>Here, we evaluated a recently developed quantification method based on Genotyping by Sequencing (GBS) for the first time in diatoms to estimate the relative abundances within a species complex. For this purpose, a reference database comprised of thousands of genomic DNA clusters was generated from cultures of Nitzschia palea. The sequencing reads from calibration and mock samples were mapped against this database for parallel quantification. We sequenced 25 mock diatom communities containing up to five taxa per sample in different abundances. Taxon abundances in these communities were also quantified by a diatom expert using manual counting of cells on light microscopic slides. The relative abundances of strains across mock samples were over- or under-estimated by the manual counting method, and a majority of mock samples had stronger correlations using GBS. Moreover, one previously recognized putative hybrid had the largest number of false positive detections demonstrating the limitation of the manual counting method when morphologically similar and/or phylogenetically close taxa are analyzed.</p><p><strong>Conclusions: </strong>Our results suggest that GBS is a reliable method to estimate the relative abundances of the N. palea taxa analyzed in this study and outperformed traditional light microscopy in terms of accuracy. GBS provides increased taxonomic resolution compared to currently available quantitative molecular approaches, and it is more scalable in the number of species that can be analyzed in a single run. Hence, this is a significant step forward in developing automated, high-throughput molecular methods specifically designed for the quantification of [diatom] communities for freshwater quality assessments.</p>\",\"PeriodicalId\":9127,\"journal\":{\"name\":\"BMC Ecology and Evolution\",\"volume\":\"23 1\",\"pages\":\"4\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2023-02-06\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9903628/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"BMC Ecology and Evolution\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1186/s12862-023-02104-2\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"\",\"JCRName\":\"\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"BMC Ecology and Evolution","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1186/s12862-023-02104-2","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"","JCRName":"","Score":null,"Total":0}
Genotyping by sequencing for estimating relative abundances of diatom taxa in mock communities.
Background: Diatoms are present in all waters and are highly sensitive to pollution gradients. Therefore, they are ideal bioindicators for water quality assessment. Current indices used in these applications are based on identifying diatom species and counting their abundances using traditional light microscopy. Several molecular techniques have been developed to help automate different steps of this process, but obtaining reliable estimates of diatom community composition and species abundance remains challenging.
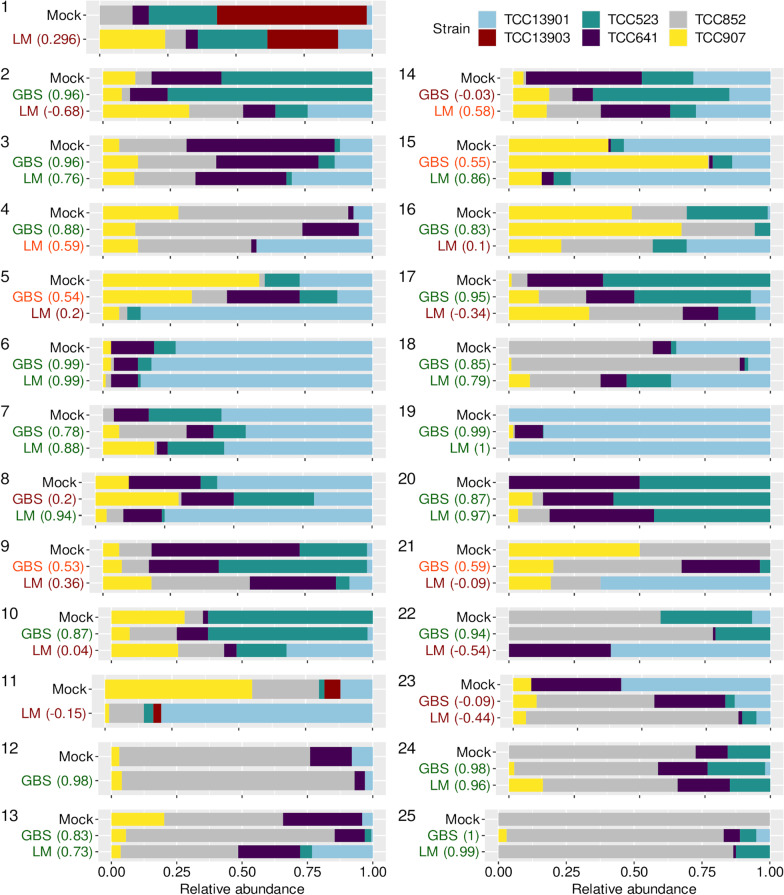
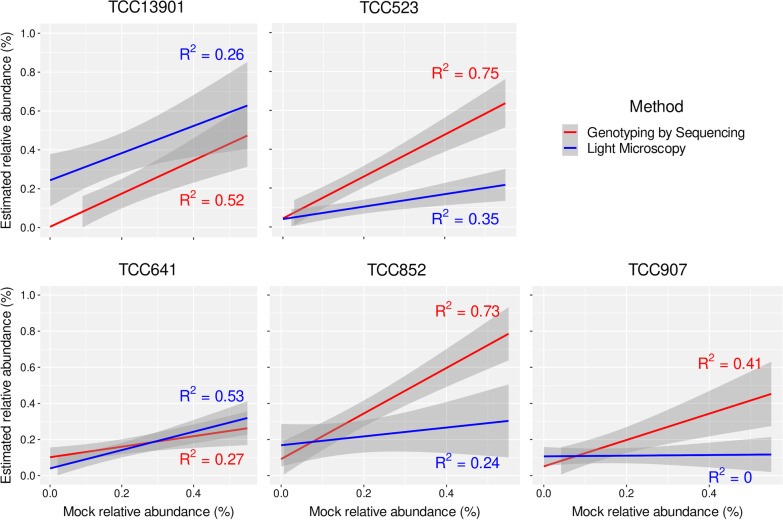
Results: Here, we evaluated a recently developed quantification method based on Genotyping by Sequencing (GBS) for the first time in diatoms to estimate the relative abundances within a species complex. For this purpose, a reference database comprised of thousands of genomic DNA clusters was generated from cultures of Nitzschia palea. The sequencing reads from calibration and mock samples were mapped against this database for parallel quantification. We sequenced 25 mock diatom communities containing up to five taxa per sample in different abundances. Taxon abundances in these communities were also quantified by a diatom expert using manual counting of cells on light microscopic slides. The relative abundances of strains across mock samples were over- or under-estimated by the manual counting method, and a majority of mock samples had stronger correlations using GBS. Moreover, one previously recognized putative hybrid had the largest number of false positive detections demonstrating the limitation of the manual counting method when morphologically similar and/or phylogenetically close taxa are analyzed.
Conclusions: Our results suggest that GBS is a reliable method to estimate the relative abundances of the N. palea taxa analyzed in this study and outperformed traditional light microscopy in terms of accuracy. GBS provides increased taxonomic resolution compared to currently available quantitative molecular approaches, and it is more scalable in the number of species that can be analyzed in a single run. Hence, this is a significant step forward in developing automated, high-throughput molecular methods specifically designed for the quantification of [diatom] communities for freshwater quality assessments.



 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
