{"title":"利用非均匀采样窗口优化计算效率的短链表面吸附平均力潜力","authors":"Naveen Kumar Vasudevan, Dongyang Li, Li Xi","doi":"10.1002/mats.202300057","DOIUrl":null,"url":null,"abstract":"<p>Free energy calculation in molecular simulation is an computationally expensive process. Umbrella sampling (US) is a go-to method for obtaining the potential of mean force (PMF) along a reaction coordinate. Its computational cost increases drastically as the molecular system gets more complex. For many polymeric and biomolecular systems, adequately sampling all configurational degrees of freedom is computationally prohibitive. Using the adsorption of a short-chain methylcellulose on a cellulose crystalline surface as the test case, this study shows that the sampling time required for reliable results is much higher than typical choices made in the literature. The accuracy of the PMF profile is strongly affected by sampling inadequacy in a few regions along the reaction coordinate. Non-uniform windows and sampling parameters are proposed to enhance the sampling in difficult regions. Sampling windows that vary with the local PMF steepness are allocated with a new algorithm. Parameters in this algorithm are optimized for the best sampling efficiency. It is demonstrated that significantly less computer time will be required to achieve the same sampling accuracy if computational resources are optimally distributed along the reaction coordinate.</p>","PeriodicalId":18157,"journal":{"name":"Macromolecular Theory and Simulations","volume":"33 1","pages":""},"PeriodicalIF":1.6000,"publicationDate":"2023-10-12","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1002/mats.202300057","citationCount":"0","resultStr":"{\"title\":\"Potential of Mean Force of Short-Chain Surface Adsorption using Non-Uniform Sampling Windows for Optimal Computational Efficiency\",\"authors\":\"Naveen Kumar Vasudevan, Dongyang Li, Li Xi\",\"doi\":\"10.1002/mats.202300057\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p>Free energy calculation in molecular simulation is an computationally expensive process. Umbrella sampling (US) is a go-to method for obtaining the potential of mean force (PMF) along a reaction coordinate. Its computational cost increases drastically as the molecular system gets more complex. For many polymeric and biomolecular systems, adequately sampling all configurational degrees of freedom is computationally prohibitive. Using the adsorption of a short-chain methylcellulose on a cellulose crystalline surface as the test case, this study shows that the sampling time required for reliable results is much higher than typical choices made in the literature. The accuracy of the PMF profile is strongly affected by sampling inadequacy in a few regions along the reaction coordinate. Non-uniform windows and sampling parameters are proposed to enhance the sampling in difficult regions. Sampling windows that vary with the local PMF steepness are allocated with a new algorithm. Parameters in this algorithm are optimized for the best sampling efficiency. It is demonstrated that significantly less computer time will be required to achieve the same sampling accuracy if computational resources are optimally distributed along the reaction coordinate.</p>\",\"PeriodicalId\":18157,\"journal\":{\"name\":\"Macromolecular Theory and Simulations\",\"volume\":\"33 1\",\"pages\":\"\"},\"PeriodicalIF\":1.6000,\"publicationDate\":\"2023-10-12\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://onlinelibrary.wiley.com/doi/epdf/10.1002/mats.202300057\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Macromolecular Theory and Simulations\",\"FirstCategoryId\":\"5\",\"ListUrlMain\":\"https://onlinelibrary.wiley.com/doi/10.1002/mats.202300057\",\"RegionNum\":4,\"RegionCategory\":\"工程技术\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q3\",\"JCRName\":\"POLYMER SCIENCE\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Macromolecular Theory and Simulations","FirstCategoryId":"5","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/mats.202300057","RegionNum":4,"RegionCategory":"工程技术","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"POLYMER SCIENCE","Score":null,"Total":0}
Potential of Mean Force of Short-Chain Surface Adsorption using Non-Uniform Sampling Windows for Optimal Computational Efficiency
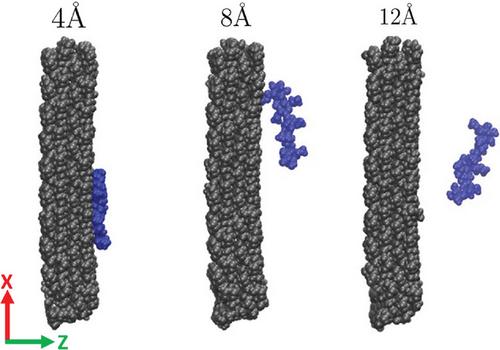
Free energy calculation in molecular simulation is an computationally expensive process. Umbrella sampling (US) is a go-to method for obtaining the potential of mean force (PMF) along a reaction coordinate. Its computational cost increases drastically as the molecular system gets more complex. For many polymeric and biomolecular systems, adequately sampling all configurational degrees of freedom is computationally prohibitive. Using the adsorption of a short-chain methylcellulose on a cellulose crystalline surface as the test case, this study shows that the sampling time required for reliable results is much higher than typical choices made in the literature. The accuracy of the PMF profile is strongly affected by sampling inadequacy in a few regions along the reaction coordinate. Non-uniform windows and sampling parameters are proposed to enhance the sampling in difficult regions. Sampling windows that vary with the local PMF steepness are allocated with a new algorithm. Parameters in this algorithm are optimized for the best sampling efficiency. It is demonstrated that significantly less computer time will be required to achieve the same sampling accuracy if computational resources are optimally distributed along the reaction coordinate.
期刊介绍:
Macromolecular Theory and Simulations is the only high-quality polymer science journal dedicated exclusively to theory and simulations, covering all aspects from macromolecular theory to advanced computer simulation techniques.





 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
