Mohsen Ranjbar, Jeremy J. Yang, Praveen Kumar, Daniel R. Byrd, Elaine L. Bearer, Tudor I. Oprea
{"title":"自噬暗基因:我们能用机器学习找到它们吗?","authors":"Mohsen Ranjbar, Jeremy J. Yang, Praveen Kumar, Daniel R. Byrd, Elaine L. Bearer, Tudor I. Oprea","doi":"10.1002/ntls.20220067","DOIUrl":null,"url":null,"abstract":"Identifying novel autophagy (ATG) associated genes in humans remains an important task for understanding this fundamental physiological process. Machine learning (ML) can highlight potentially “missing pieces” linking core ATG genes with understudied, “dark” genes by mining functional genomic data. Here, a set of 103 (out of 288 genes from the Autophagy Database) was used as training set, based on ATG-associated terms annotated from 3 secondary sources: GO (gene ontology), Kyoto Encyclopedia of Genes and Genomes pathway, and UniProt keywords, as additional confirmation of their importance in ATG. As negative labels, an OMIM list of genes associated with monogenic diseases was used (after excluding the 288 ATG-associated genes). Data related to these genes from 17 different sources were compiled and used to derive a trained MetaPath/XGBoost (MPxgb) ML model for distinguishing ATG and non-ATG genes (10-fold cross-validated, 100-times randomized models, median area under the curve = 0.994 ± 0.008). Sixteen ATG-relevant variables explained 64% of the total model gain. Overall, 23% of the top 251 predicted genes are annotated in the Autophagy Database, whereas 193 genes (77%) are not. In 2019, we suggested that some of these 193 genes may represent “ATG dark genes.” A literature search in 2022 for those top 20 predicted ATG dark genes found that 9 were subsequently reported as ATG genes during the intervening 3.5 years. A post-factum evaluation of data leakage (the presence of ATG-associated terms in the top 40 ML features) confirms that 7 out of these 9 genes and 2 out of 3 other recently validated predictions from the bottom 20 are novel. Those genes with the largest number of ATG features would be most likely to yield valuable experimental insights. Modern high-throughput testing would be capable of spanning the full 193 ATG genes list reported here. Our analysis demonstrates that ML can guide genomics research to gain a more complete functional and pathway annotation of complex processes. Key points – A knowledge-graph based machine learning model was designed for predicting unknown autophagy genes via mining functional genomic data. – Literature search validated predicted genes. – Our machine learning models could be generalized and applied to other genomic libraries to uncover dark genes for various functions.","PeriodicalId":74244,"journal":{"name":"Natural sciences (Weinheim, Germany)","volume":"22 1","pages":"0"},"PeriodicalIF":3.1000,"publicationDate":"2023-05-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Autophagy dark genes: Can we find them with machine learning?\",\"authors\":\"Mohsen Ranjbar, Jeremy J. Yang, Praveen Kumar, Daniel R. Byrd, Elaine L. Bearer, Tudor I. Oprea\",\"doi\":\"10.1002/ntls.20220067\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"Identifying novel autophagy (ATG) associated genes in humans remains an important task for understanding this fundamental physiological process. Machine learning (ML) can highlight potentially “missing pieces” linking core ATG genes with understudied, “dark” genes by mining functional genomic data. Here, a set of 103 (out of 288 genes from the Autophagy Database) was used as training set, based on ATG-associated terms annotated from 3 secondary sources: GO (gene ontology), Kyoto Encyclopedia of Genes and Genomes pathway, and UniProt keywords, as additional confirmation of their importance in ATG. As negative labels, an OMIM list of genes associated with monogenic diseases was used (after excluding the 288 ATG-associated genes). Data related to these genes from 17 different sources were compiled and used to derive a trained MetaPath/XGBoost (MPxgb) ML model for distinguishing ATG and non-ATG genes (10-fold cross-validated, 100-times randomized models, median area under the curve = 0.994 ± 0.008). Sixteen ATG-relevant variables explained 64% of the total model gain. Overall, 23% of the top 251 predicted genes are annotated in the Autophagy Database, whereas 193 genes (77%) are not. In 2019, we suggested that some of these 193 genes may represent “ATG dark genes.” A literature search in 2022 for those top 20 predicted ATG dark genes found that 9 were subsequently reported as ATG genes during the intervening 3.5 years. A post-factum evaluation of data leakage (the presence of ATG-associated terms in the top 40 ML features) confirms that 7 out of these 9 genes and 2 out of 3 other recently validated predictions from the bottom 20 are novel. Those genes with the largest number of ATG features would be most likely to yield valuable experimental insights. Modern high-throughput testing would be capable of spanning the full 193 ATG genes list reported here. Our analysis demonstrates that ML can guide genomics research to gain a more complete functional and pathway annotation of complex processes. Key points – A knowledge-graph based machine learning model was designed for predicting unknown autophagy genes via mining functional genomic data. – Literature search validated predicted genes. – Our machine learning models could be generalized and applied to other genomic libraries to uncover dark genes for various functions.\",\"PeriodicalId\":74244,\"journal\":{\"name\":\"Natural sciences (Weinheim, Germany)\",\"volume\":\"22 1\",\"pages\":\"0\"},\"PeriodicalIF\":3.1000,\"publicationDate\":\"2023-05-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Natural sciences (Weinheim, Germany)\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1002/ntls.20220067\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"MULTIDISCIPLINARY SCIENCES\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Natural sciences (Weinheim, Germany)","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1002/ntls.20220067","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"MULTIDISCIPLINARY SCIENCES","Score":null,"Total":0}
Autophagy dark genes: Can we find them with machine learning?
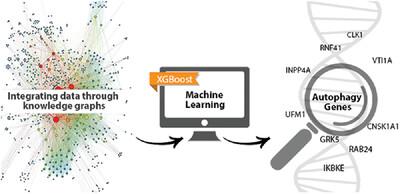
Identifying novel autophagy (ATG) associated genes in humans remains an important task for understanding this fundamental physiological process. Machine learning (ML) can highlight potentially “missing pieces” linking core ATG genes with understudied, “dark” genes by mining functional genomic data. Here, a set of 103 (out of 288 genes from the Autophagy Database) was used as training set, based on ATG-associated terms annotated from 3 secondary sources: GO (gene ontology), Kyoto Encyclopedia of Genes and Genomes pathway, and UniProt keywords, as additional confirmation of their importance in ATG. As negative labels, an OMIM list of genes associated with monogenic diseases was used (after excluding the 288 ATG-associated genes). Data related to these genes from 17 different sources were compiled and used to derive a trained MetaPath/XGBoost (MPxgb) ML model for distinguishing ATG and non-ATG genes (10-fold cross-validated, 100-times randomized models, median area under the curve = 0.994 ± 0.008). Sixteen ATG-relevant variables explained 64% of the total model gain. Overall, 23% of the top 251 predicted genes are annotated in the Autophagy Database, whereas 193 genes (77%) are not. In 2019, we suggested that some of these 193 genes may represent “ATG dark genes.” A literature search in 2022 for those top 20 predicted ATG dark genes found that 9 were subsequently reported as ATG genes during the intervening 3.5 years. A post-factum evaluation of data leakage (the presence of ATG-associated terms in the top 40 ML features) confirms that 7 out of these 9 genes and 2 out of 3 other recently validated predictions from the bottom 20 are novel. Those genes with the largest number of ATG features would be most likely to yield valuable experimental insights. Modern high-throughput testing would be capable of spanning the full 193 ATG genes list reported here. Our analysis demonstrates that ML can guide genomics research to gain a more complete functional and pathway annotation of complex processes. Key points – A knowledge-graph based machine learning model was designed for predicting unknown autophagy genes via mining functional genomic data. – Literature search validated predicted genes. – Our machine learning models could be generalized and applied to other genomic libraries to uncover dark genes for various functions.


 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
