{"title":"新型错义变异导致slc5a6相关疾病表型谱中的中间表型。","authors":"Yasuhiro Utsuno, Keisuke Hamada, Kohei Hamanaka, Keita Miyoshi, Keiji Tsuchimoto, Satoshi Sunada, Toshiyuki Itai, Masamune Sakamoto, Naomi Tsuchida, Yuri Uchiyama, Eriko Koshimizu, Atsushi Fujita, Satoko Miyatake, Kazuharu Misawa, Takeshi Mizuguchi, Yasuhito Kato, Kuniaki Saito, Kazuhiro Ogata, Naomichi Matsumoto","doi":"10.1038/s10038-023-01206-5","DOIUrl":null,"url":null,"abstract":"SLC5A6 encodes the sodium-dependent multivitamin transporter, a transmembrane protein that uptakes biotin, pantothenic acid, and lipoic acid. Biallelic SLC5A6 variants cause sodium-dependent multivitamin transporter deficiency (SMVTD) and childhood-onset biotin-responsive peripheral motor neuropathy (COMNB), which both respond well to replacement therapy with the above three nutrients. SMVTD usually presents with various symptoms in multiple organs, such as gastrointestinal hemorrhage, brain atrophy, and global developmental delay, at birth or in infancy. Without nutrient replacement therapy, SMVTD can be lethal in early childhood. COMNB is clinically milder and has a later onset than SMVTD, at approximately 10 years of age. COMNB symptoms are mostly limited to peripheral motor neuropathy. Here we report three patients from one Japanese family harboring novel compound heterozygous missense variants in SLC5A6, namely NM_021095.4:c.[221C>T];[642G>C] p.[(Ser74Phe)];[(Gln214His)]. Both variants were predicted to be deleterious through multiple lines of evidence, including amino acid conservation, in silico predictions of pathogenicity, and protein structure considerations. Drosophila analysis also showed c.221C>T to be pathogenic. All three patients had congenital brain cysts on neonatal cranial imaging, but no other morphological abnormalities. They also had a mild motor developmental delay that almost completely resolved despite no treatment. In terms of severity, their phenotypes were intermediate between SMVTD and COMNB. From these findings we propose a new SLC5A6-related disorder, spontaneously remitting developmental delay with brain cysts (SRDDBC) whose phenotypic severity is between that of SMVTD and COMNB. Further clinical and genetic evidence is needed to support our suggestion.","PeriodicalId":16077,"journal":{"name":"Journal of Human Genetics","volume":"69 2","pages":"69-77"},"PeriodicalIF":2.5000,"publicationDate":"2023-11-27","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Novel missense variants cause intermediate phenotypes in the phenotypic spectrum of SLC5A6-related disorders\",\"authors\":\"Yasuhiro Utsuno, Keisuke Hamada, Kohei Hamanaka, Keita Miyoshi, Keiji Tsuchimoto, Satoshi Sunada, Toshiyuki Itai, Masamune Sakamoto, Naomi Tsuchida, Yuri Uchiyama, Eriko Koshimizu, Atsushi Fujita, Satoko Miyatake, Kazuharu Misawa, Takeshi Mizuguchi, Yasuhito Kato, Kuniaki Saito, Kazuhiro Ogata, Naomichi Matsumoto\",\"doi\":\"10.1038/s10038-023-01206-5\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"SLC5A6 encodes the sodium-dependent multivitamin transporter, a transmembrane protein that uptakes biotin, pantothenic acid, and lipoic acid. Biallelic SLC5A6 variants cause sodium-dependent multivitamin transporter deficiency (SMVTD) and childhood-onset biotin-responsive peripheral motor neuropathy (COMNB), which both respond well to replacement therapy with the above three nutrients. SMVTD usually presents with various symptoms in multiple organs, such as gastrointestinal hemorrhage, brain atrophy, and global developmental delay, at birth or in infancy. Without nutrient replacement therapy, SMVTD can be lethal in early childhood. COMNB is clinically milder and has a later onset than SMVTD, at approximately 10 years of age. COMNB symptoms are mostly limited to peripheral motor neuropathy. Here we report three patients from one Japanese family harboring novel compound heterozygous missense variants in SLC5A6, namely NM_021095.4:c.[221C>T];[642G>C] p.[(Ser74Phe)];[(Gln214His)]. Both variants were predicted to be deleterious through multiple lines of evidence, including amino acid conservation, in silico predictions of pathogenicity, and protein structure considerations. Drosophila analysis also showed c.221C>T to be pathogenic. All three patients had congenital brain cysts on neonatal cranial imaging, but no other morphological abnormalities. They also had a mild motor developmental delay that almost completely resolved despite no treatment. In terms of severity, their phenotypes were intermediate between SMVTD and COMNB. From these findings we propose a new SLC5A6-related disorder, spontaneously remitting developmental delay with brain cysts (SRDDBC) whose phenotypic severity is between that of SMVTD and COMNB. Further clinical and genetic evidence is needed to support our suggestion.\",\"PeriodicalId\":16077,\"journal\":{\"name\":\"Journal of Human Genetics\",\"volume\":\"69 2\",\"pages\":\"69-77\"},\"PeriodicalIF\":2.5000,\"publicationDate\":\"2023-11-27\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Human Genetics\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://www.nature.com/articles/s10038-023-01206-5\",\"RegionNum\":3,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"GENETICS & HEREDITY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Human Genetics","FirstCategoryId":"99","ListUrlMain":"https://www.nature.com/articles/s10038-023-01206-5","RegionNum":3,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
Novel missense variants cause intermediate phenotypes in the phenotypic spectrum of SLC5A6-related disorders
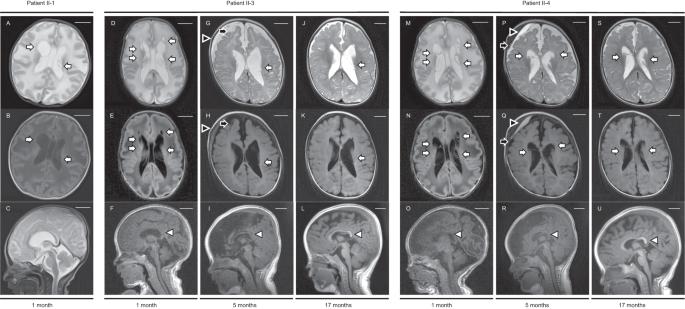
SLC5A6 encodes the sodium-dependent multivitamin transporter, a transmembrane protein that uptakes biotin, pantothenic acid, and lipoic acid. Biallelic SLC5A6 variants cause sodium-dependent multivitamin transporter deficiency (SMVTD) and childhood-onset biotin-responsive peripheral motor neuropathy (COMNB), which both respond well to replacement therapy with the above three nutrients. SMVTD usually presents with various symptoms in multiple organs, such as gastrointestinal hemorrhage, brain atrophy, and global developmental delay, at birth or in infancy. Without nutrient replacement therapy, SMVTD can be lethal in early childhood. COMNB is clinically milder and has a later onset than SMVTD, at approximately 10 years of age. COMNB symptoms are mostly limited to peripheral motor neuropathy. Here we report three patients from one Japanese family harboring novel compound heterozygous missense variants in SLC5A6, namely NM_021095.4:c.[221C>T];[642G>C] p.[(Ser74Phe)];[(Gln214His)]. Both variants were predicted to be deleterious through multiple lines of evidence, including amino acid conservation, in silico predictions of pathogenicity, and protein structure considerations. Drosophila analysis also showed c.221C>T to be pathogenic. All three patients had congenital brain cysts on neonatal cranial imaging, but no other morphological abnormalities. They also had a mild motor developmental delay that almost completely resolved despite no treatment. In terms of severity, their phenotypes were intermediate between SMVTD and COMNB. From these findings we propose a new SLC5A6-related disorder, spontaneously remitting developmental delay with brain cysts (SRDDBC) whose phenotypic severity is between that of SMVTD and COMNB. Further clinical and genetic evidence is needed to support our suggestion.
期刊介绍:
The Journal of Human Genetics is an international journal publishing articles on human genetics, including medical genetics and human genome analysis. It covers all aspects of human genetics, including molecular genetics, clinical genetics, behavioral genetics, immunogenetics, pharmacogenomics, population genetics, functional genomics, epigenetics, genetic counseling and gene therapy.
Articles on the following areas are especially welcome: genetic factors of monogenic and complex disorders, genome-wide association studies, genetic epidemiology, cancer genetics, personal genomics, genotype-phenotype relationships and genome diversity.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
