Nicola Ferri, Sara De Martin, James Stuart, Sergio Traversa, Franco Folli, Marco Pappagallo, Cedric O'Gorman, Clotilde Guidetti, Andrea Mattarei, Charles E Inturrisi, Paolo L Manfredi
{"title":"艾美沙酮(REL-1017)参与CYP3A4-和cyp2d6介导代谢的药物相互作用研究。","authors":"Nicola Ferri, Sara De Martin, James Stuart, Sergio Traversa, Franco Folli, Marco Pappagallo, Cedric O'Gorman, Clotilde Guidetti, Andrea Mattarei, Charles E Inturrisi, Paolo L Manfredi","doi":"10.1007/s40268-023-00450-6","DOIUrl":null,"url":null,"abstract":"<p><strong>Background and objective: </strong>Esmethadone (dextromethadone; d-methadone; S-methadone (+)-methadone; REL-1017) is the opioid inactive dextro-isomer of racemic methadone. Esmethadone is a low potency N-methyl-D-aspartate (NMDA) receptor channel blocker with higher affinity for GluN2D subtypes. Esmethadone showed robust, rapid, and sustained antidepressant effects in patients with major depressive disorder (MDD) with inadequate response to ongoing serotonergic antidepressant treatment.</p><p><strong>Methods: </strong>Here we described the results of in vitro and phase 1 clinical trials aimed at investigating the esmethadone metabolism and possible drug-drug interactions.</p><p><strong>Results: </strong>Esmethadone is primarily metabolized to EDDP (2-ethylene-1,5-dimethyl-3,3-diphenylpyrrolidine) by multiple enzymes, including CYP3A4/5 and CYP2B6. In vitro studies showed that esmethadone inhibits CYP2D6 with IC<sub>50</sub> of 9.6 μM and is an inducer of CYP3A4/5. The clinical relevance of the inhibition of CYP2D6 and the induction of CYP3A4 were investigated by co-administering esmethadone and dextromethorphan (a substrate for CYP2D6) or midazolam (a substrate for CYP3A4) in healthy volunteers. The administration of esmethadone at the dosage of 75 mg (which is the loading dose administered to patients in MDD clinical trials) significantly increased the exposure (AUC) of both dextromethorphan and its metabolite dextrorphan by 2.71 and 3.11-fold, respectively. Esmethadone did not modify the pharmacokinetic profile of midazolam, while it increased C<sub>max</sub> and AUC of its metabolite 1'-hydroxymidazolam by 2.4- and 3.8-fold, respectively. A second study evaluated the effect of the CYP3A4 inhibitor cobicistat on the pharmacokinetics of esmethadone. Cobicistat slightly increase (+32%) the total exposure (AUC<sub>0-inf</sub>) of esmethadone.</p><p><strong>Conclusions: </strong>In summary, esmethadone demonstrated a negligible effect on CYP3A4 induction and its metabolism was not meaningfully affected by strong CYP3A4 inhibitors while it increased exposure of CYP2D6-metabolized drugs.</p>","PeriodicalId":49258,"journal":{"name":"Drugs in Research & Development","volume":" ","pages":"51-68"},"PeriodicalIF":2.1000,"publicationDate":"2024-03-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11035515/pdf/","citationCount":"0","resultStr":"{\"title\":\"Drug-Drug Interaction Studies of Esmethadone (REL-1017) Involving CYP3A4- and CYP2D6-Mediated Metabolism.\",\"authors\":\"Nicola Ferri, Sara De Martin, James Stuart, Sergio Traversa, Franco Folli, Marco Pappagallo, Cedric O'Gorman, Clotilde Guidetti, Andrea Mattarei, Charles E Inturrisi, Paolo L Manfredi\",\"doi\":\"10.1007/s40268-023-00450-6\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Background and objective: </strong>Esmethadone (dextromethadone; d-methadone; S-methadone (+)-methadone; REL-1017) is the opioid inactive dextro-isomer of racemic methadone. Esmethadone is a low potency N-methyl-D-aspartate (NMDA) receptor channel blocker with higher affinity for GluN2D subtypes. Esmethadone showed robust, rapid, and sustained antidepressant effects in patients with major depressive disorder (MDD) with inadequate response to ongoing serotonergic antidepressant treatment.</p><p><strong>Methods: </strong>Here we described the results of in vitro and phase 1 clinical trials aimed at investigating the esmethadone metabolism and possible drug-drug interactions.</p><p><strong>Results: </strong>Esmethadone is primarily metabolized to EDDP (2-ethylene-1,5-dimethyl-3,3-diphenylpyrrolidine) by multiple enzymes, including CYP3A4/5 and CYP2B6. In vitro studies showed that esmethadone inhibits CYP2D6 with IC<sub>50</sub> of 9.6 μM and is an inducer of CYP3A4/5. The clinical relevance of the inhibition of CYP2D6 and the induction of CYP3A4 were investigated by co-administering esmethadone and dextromethorphan (a substrate for CYP2D6) or midazolam (a substrate for CYP3A4) in healthy volunteers. The administration of esmethadone at the dosage of 75 mg (which is the loading dose administered to patients in MDD clinical trials) significantly increased the exposure (AUC) of both dextromethorphan and its metabolite dextrorphan by 2.71 and 3.11-fold, respectively. Esmethadone did not modify the pharmacokinetic profile of midazolam, while it increased C<sub>max</sub> and AUC of its metabolite 1'-hydroxymidazolam by 2.4- and 3.8-fold, respectively. A second study evaluated the effect of the CYP3A4 inhibitor cobicistat on the pharmacokinetics of esmethadone. Cobicistat slightly increase (+32%) the total exposure (AUC<sub>0-inf</sub>) of esmethadone.</p><p><strong>Conclusions: </strong>In summary, esmethadone demonstrated a negligible effect on CYP3A4 induction and its metabolism was not meaningfully affected by strong CYP3A4 inhibitors while it increased exposure of CYP2D6-metabolized drugs.</p>\",\"PeriodicalId\":49258,\"journal\":{\"name\":\"Drugs in Research & Development\",\"volume\":\" \",\"pages\":\"51-68\"},\"PeriodicalIF\":2.1000,\"publicationDate\":\"2024-03-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11035515/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Drugs in Research & Development\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://doi.org/10.1007/s40268-023-00450-6\",\"RegionNum\":4,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2023/11/27 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"Q3\",\"JCRName\":\"PHARMACOLOGY & PHARMACY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Drugs in Research & Development","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1007/s40268-023-00450-6","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2023/11/27 0:00:00","PubModel":"Epub","JCR":"Q3","JCRName":"PHARMACOLOGY & PHARMACY","Score":null,"Total":0}
引用次数: 0
摘要
背景与目的:艾美沙酮(右美沙酮;d-methadone;S-methadone(+)美沙酮;REL-1017)是外消旋美沙酮的阿片非活性右旋异构体。艾美沙酮是一种低效n -甲基- d -天冬氨酸(NMDA)受体通道阻滞剂,对GluN2D亚型具有较高的亲和力。艾美沙酮对重度抑郁症(MDD)患者表现出强劲、快速和持续的抗抑郁作用,这些患者对持续的5 -羟色胺能抗抑郁治疗反应不足。方法:本文描述了旨在研究艾美沙酮代谢和可能的药物-药物相互作用的体外和1期临床试验的结果。结果:艾美沙酮主要通过多种酶代谢生成EDDP(2-乙烯-1,5-二甲基-3,3-二苯基吡咯烷),包括CYP3A4/5和CYP2B6。体外研究表明,艾美沙酮抑制CYP2D6的IC50为9.6 μM,是CYP3A4/5的诱诱剂。通过在健康志愿者中联合使用艾美沙酮和右美沙芬(CYP2D6的底物)或咪达唑仑(CYP3A4的底物)来研究抑制CYP2D6和诱导CYP3A4的临床相关性。以75 mg的剂量(MDD临床试验中给予患者的负荷剂量)施用艾美沙酮,右美沙芬及其代谢物右美沙芬的暴露量(AUC)分别显著增加2.71倍和3.11倍。艾美沙酮没有改变咪达唑仑的药动学特征,但使其代谢物1′-羟咪达唑仑的Cmax和AUC分别增加了2.4倍和3.8倍。第二项研究评估了CYP3A4抑制剂cobicistat对艾美沙酮药代动力学的影响。可比司他轻微增加艾美沙酮的总暴露量(AUC0-inf)(+32%)。结论:综上所述,艾美沙酮对CYP3A4的诱导作用可以忽略不计,强CYP3A4抑制剂对其代谢没有显著影响,但增加了cyp2d6代谢药物的暴露。
Drug-Drug Interaction Studies of Esmethadone (REL-1017) Involving CYP3A4- and CYP2D6-Mediated Metabolism.
Background and objective: Esmethadone (dextromethadone; d-methadone; S-methadone (+)-methadone; REL-1017) is the opioid inactive dextro-isomer of racemic methadone. Esmethadone is a low potency N-methyl-D-aspartate (NMDA) receptor channel blocker with higher affinity for GluN2D subtypes. Esmethadone showed robust, rapid, and sustained antidepressant effects in patients with major depressive disorder (MDD) with inadequate response to ongoing serotonergic antidepressant treatment.
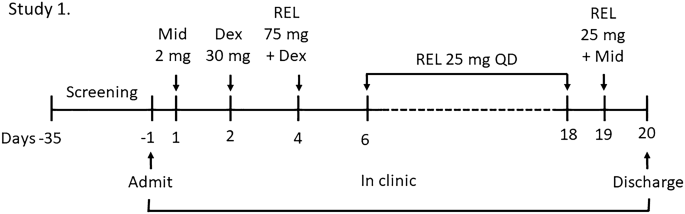
Methods: Here we described the results of in vitro and phase 1 clinical trials aimed at investigating the esmethadone metabolism and possible drug-drug interactions.
Results: Esmethadone is primarily metabolized to EDDP (2-ethylene-1,5-dimethyl-3,3-diphenylpyrrolidine) by multiple enzymes, including CYP3A4/5 and CYP2B6. In vitro studies showed that esmethadone inhibits CYP2D6 with IC50 of 9.6 μM and is an inducer of CYP3A4/5. The clinical relevance of the inhibition of CYP2D6 and the induction of CYP3A4 were investigated by co-administering esmethadone and dextromethorphan (a substrate for CYP2D6) or midazolam (a substrate for CYP3A4) in healthy volunteers. The administration of esmethadone at the dosage of 75 mg (which is the loading dose administered to patients in MDD clinical trials) significantly increased the exposure (AUC) of both dextromethorphan and its metabolite dextrorphan by 2.71 and 3.11-fold, respectively. Esmethadone did not modify the pharmacokinetic profile of midazolam, while it increased Cmax and AUC of its metabolite 1'-hydroxymidazolam by 2.4- and 3.8-fold, respectively. A second study evaluated the effect of the CYP3A4 inhibitor cobicistat on the pharmacokinetics of esmethadone. Cobicistat slightly increase (+32%) the total exposure (AUC0-inf) of esmethadone.
Conclusions: In summary, esmethadone demonstrated a negligible effect on CYP3A4 induction and its metabolism was not meaningfully affected by strong CYP3A4 inhibitors while it increased exposure of CYP2D6-metabolized drugs.
期刊介绍:
Drugs in R&D is an international, peer reviewed, open access, online only journal, and provides timely information from all phases of drug research and development that will inform clinical practice. Healthcare decision makers are thus provided with knowledge about the developing place of a drug in therapy.
The Journal includes:
Clinical research on new and established drugs;
Preclinical research of direct relevance to clinical drug development;
Short communications and case study reports that meet the above criteria will also be considered;
Reviews may also be considered.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
