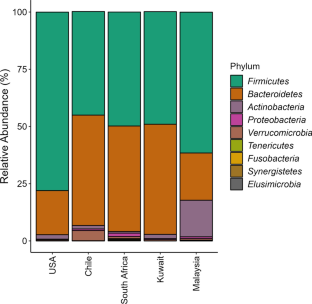
{"title":"探索地理差异对人类肠道微生物组序列特征的影响","authors":"Gauraw Kumar, Punyasloke Bhadury","doi":"10.1007/s12041-023-01448-4","DOIUrl":null,"url":null,"abstract":"<p>Geography shapes the structure and function of human gut microbiomes. In this study, we have explored the available human gut microbiome 16S rRNA sequence datasets of cohorts representing large geographical gradients. The 16S rRNA sequences representing V3-V4 as well as V4 regions generated using Illumina sequencing chemistry in the MiSeq platform encompassing the United States of America, Chile, South Africa, Kuwait, and Malaysia were subjected to in-depth computational biology analyses. Firmicutes and Bacteroidetes were the most dominant phyla present in all studied cohorts but Actinobacteria was exclusively present in high abundance in cohorts from Malaysia (15.99%). The relative abundance of five families, namely Bacteroidaceae, Ruminococcaceae, Prevotellaceae, Clostridiaceae, and Eubacteriaceae were highest representing the studied cohorts. The permutational multivariate analysis of variance (PERMANOVA) showed that the dissimilarity in the gut microbiome structure of cohorts representing studied countries was significant (R<sup>2</sup> = 0.28, <i>P</i> < 0.001). The calculated Firmicutes to Bacteroidetes (F : B) ratio was found to be lowest in cohorts from South Africa (1.11) and Chile (0.95). The cohorts from South Africa exhibited the highest alpha diversity based on Hill numbers at <i>q</i>=0, whereas at <i>q</i>=1 and 2, cohorts from Malaysia had the highest alpha diversity. The beta diversity analysis revealed that cohorts from Chile formed a distinct cluster among all the studied geographical locations. For the first time, the study also showed that cohorts from Malaysia representing short geographical distances exhibited distinct intrapopulation differences in the gut microbiome and may not be influenced by cultural and genetic factors.</p>","PeriodicalId":15907,"journal":{"name":"Journal of Genetics","volume":"346 2","pages":""},"PeriodicalIF":1.2000,"publicationDate":"2023-11-16","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Exploring the influences of geographical variation on sequence signatures in the human gut microbiome\",\"authors\":\"Gauraw Kumar, Punyasloke Bhadury\",\"doi\":\"10.1007/s12041-023-01448-4\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p>Geography shapes the structure and function of human gut microbiomes. In this study, we have explored the available human gut microbiome 16S rRNA sequence datasets of cohorts representing large geographical gradients. The 16S rRNA sequences representing V3-V4 as well as V4 regions generated using Illumina sequencing chemistry in the MiSeq platform encompassing the United States of America, Chile, South Africa, Kuwait, and Malaysia were subjected to in-depth computational biology analyses. Firmicutes and Bacteroidetes were the most dominant phyla present in all studied cohorts but Actinobacteria was exclusively present in high abundance in cohorts from Malaysia (15.99%). The relative abundance of five families, namely Bacteroidaceae, Ruminococcaceae, Prevotellaceae, Clostridiaceae, and Eubacteriaceae were highest representing the studied cohorts. The permutational multivariate analysis of variance (PERMANOVA) showed that the dissimilarity in the gut microbiome structure of cohorts representing studied countries was significant (R<sup>2</sup> = 0.28, <i>P</i> < 0.001). The calculated Firmicutes to Bacteroidetes (F : B) ratio was found to be lowest in cohorts from South Africa (1.11) and Chile (0.95). The cohorts from South Africa exhibited the highest alpha diversity based on Hill numbers at <i>q</i>=0, whereas at <i>q</i>=1 and 2, cohorts from Malaysia had the highest alpha diversity. The beta diversity analysis revealed that cohorts from Chile formed a distinct cluster among all the studied geographical locations. For the first time, the study also showed that cohorts from Malaysia representing short geographical distances exhibited distinct intrapopulation differences in the gut microbiome and may not be influenced by cultural and genetic factors.</p>\",\"PeriodicalId\":15907,\"journal\":{\"name\":\"Journal of Genetics\",\"volume\":\"346 2\",\"pages\":\"\"},\"PeriodicalIF\":1.2000,\"publicationDate\":\"2023-11-16\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Genetics\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://doi.org/10.1007/s12041-023-01448-4\",\"RegionNum\":4,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"EDUCATION & EDUCATIONAL RESEARCH\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Genetics","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1007/s12041-023-01448-4","RegionNum":4,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"EDUCATION & EDUCATIONAL RESEARCH","Score":null,"Total":0}
引用次数: 0
摘要
地理位置决定了人类肠道微生物群的结构和功能。在这项研究中,我们探索了具有大地理梯度的队列中可用的人类肠道微生物组16S rRNA序列数据集。在美国、智利、南非、科威特和马来西亚的MiSeq平台上使用Illumina测序化学生成的代表V3-V4以及V4区域的16S rRNA序列进行了深入的计算生物学分析。厚壁菌门和拟杆菌门是所有研究队列中最主要的门,但放线菌门在马来西亚队列中仅以高丰度存在(15.99%)。Bacteroidaceae、Ruminococcaceae、Prevotellaceae、Clostridiaceae和Eubacteriaceae这5个科的相对丰度在研究队列中最高。排列多变量方差分析(PERMANOVA)显示,代表研究国家的队列肠道微生物群结构存在显著差异(R2 = 0.28, P <0.001)。计算得出的厚壁菌门与拟杆菌门(F: B)比值在南非(1.11)和智利(0.95)的队列中最低。在q=0时,南非群体表现出最高的α多样性,而在q=1和2时,马来西亚群体表现出最高的α多样性。beta多样性分析显示,在所有研究的地理位置中,来自智利的队列形成了一个独特的集群。该研究还首次表明,来自马来西亚的地理距离较短的人群在肠道微生物组中表现出明显的种群内差异,可能不受文化和遗传因素的影响。
Exploring the influences of geographical variation on sequence signatures in the human gut microbiome
Geography shapes the structure and function of human gut microbiomes. In this study, we have explored the available human gut microbiome 16S rRNA sequence datasets of cohorts representing large geographical gradients. The 16S rRNA sequences representing V3-V4 as well as V4 regions generated using Illumina sequencing chemistry in the MiSeq platform encompassing the United States of America, Chile, South Africa, Kuwait, and Malaysia were subjected to in-depth computational biology analyses. Firmicutes and Bacteroidetes were the most dominant phyla present in all studied cohorts but Actinobacteria was exclusively present in high abundance in cohorts from Malaysia (15.99%). The relative abundance of five families, namely Bacteroidaceae, Ruminococcaceae, Prevotellaceae, Clostridiaceae, and Eubacteriaceae were highest representing the studied cohorts. The permutational multivariate analysis of variance (PERMANOVA) showed that the dissimilarity in the gut microbiome structure of cohorts representing studied countries was significant (R2 = 0.28, P < 0.001). The calculated Firmicutes to Bacteroidetes (F : B) ratio was found to be lowest in cohorts from South Africa (1.11) and Chile (0.95). The cohorts from South Africa exhibited the highest alpha diversity based on Hill numbers at q=0, whereas at q=1 and 2, cohorts from Malaysia had the highest alpha diversity. The beta diversity analysis revealed that cohorts from Chile formed a distinct cluster among all the studied geographical locations. For the first time, the study also showed that cohorts from Malaysia representing short geographical distances exhibited distinct intrapopulation differences in the gut microbiome and may not be influenced by cultural and genetic factors.
期刊介绍:
The journal retains its traditional interest in evolutionary research that is of relevance to geneticists, even if this is not explicitly genetical in nature. The journal covers all areas of genetics and evolution,including molecular genetics and molecular evolution.It publishes papers and review articles on current topics, commentaries and essayson ideas and trends in genetics and evolutionary biology, historical developments, debates and book reviews. From 2010 onwards, the journal has published a special category of papers termed ‘Online Resources’. These are brief reports on the development and the routine use of molecular markers for assessing genetic variability within and among species. Also published are reports outlining pedagogical approaches in genetics teaching.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
