Haiyang Duan, Jianxin Li, Li Sun, Xuehang Xiong, Shuhao Xu, Yan Sun, Xiaolong Ju, Zhengjie Xue, Jionghao Gao, Yan Wang, Huiling Xie, Dong Ding, Xuehai Zhang, Jihua Tang
{"title":"通过使用扩大的 SNP 面板进行全基因组关联研究,确定与玉米籽粒中淀粉含量相关的新位点","authors":"Haiyang Duan, Jianxin Li, Li Sun, Xuehang Xiong, Shuhao Xu, Yan Sun, Xiaolong Ju, Zhengjie Xue, Jionghao Gao, Yan Wang, Huiling Xie, Dong Ding, Xuehai Zhang, Jihua Tang","doi":"10.1007/s11032-023-01437-6","DOIUrl":null,"url":null,"abstract":"<p>Starch is a major component of cereals, comprising over 70% of dry weight. It serves as a primary carbon source for humans and animals. In addition, starch is an indispensable industrial raw material. While maize (<i>Zea mays</i>) is a key crop and the primary source of starch, the genetic basis for starch content in maize kernels remains poorly understood. In this study, using an enlarged panel, we conducted a genome-wide association study (GWAS) based on best linear unbiased prediction (BLUP) value for starch content of 261 inbred lines across three environments. Compared with previous study, we identified 14 additional significant quantitative trait loci (QTL), encompassed a total of 42 genes, and indicated that increased marker density contributes to improved statistical power. By integrating gene expression profiling, Gene Ontology (GO) enrichment and haplotype analysis, several potential target genes that may play a role in regulating starch content in maize kernels have been identified. Notably, we found that <i>ZmAPC4</i>, associated with the significant SNP chr4.S_175584318, which encodes a WD40 repeat-like superfamily protein and is highly expressed in maize endosperm, might be a crucial regulator of maize kernel starch synthesis. Out of the 261 inbred lines analyzed, they were categorized into four haplotypes. Remarkably, it was observed that the inbred lines harboring hap4 demonstrated the highest starch content compared to the other haplotypes. Additionally, as a significant achievement, we have developed molecular markers that effectively differentiate maize inbred lines based on their starch content. Overall, our study provides valuable insights into the genetic basis of starch content and the molecular markers can be useful in breeding programs aimed at developing maize varieties with high starch content, thereby improving breeding efficiency.</p>","PeriodicalId":18769,"journal":{"name":"Molecular Breeding","volume":"19 1","pages":""},"PeriodicalIF":3.0000,"publicationDate":"2023-12-12","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Identification of novel loci associated with starch content in maize kernels by a genome-wide association study using an enlarged SNP panel\",\"authors\":\"Haiyang Duan, Jianxin Li, Li Sun, Xuehang Xiong, Shuhao Xu, Yan Sun, Xiaolong Ju, Zhengjie Xue, Jionghao Gao, Yan Wang, Huiling Xie, Dong Ding, Xuehai Zhang, Jihua Tang\",\"doi\":\"10.1007/s11032-023-01437-6\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p>Starch is a major component of cereals, comprising over 70% of dry weight. It serves as a primary carbon source for humans and animals. In addition, starch is an indispensable industrial raw material. While maize (<i>Zea mays</i>) is a key crop and the primary source of starch, the genetic basis for starch content in maize kernels remains poorly understood. In this study, using an enlarged panel, we conducted a genome-wide association study (GWAS) based on best linear unbiased prediction (BLUP) value for starch content of 261 inbred lines across three environments. Compared with previous study, we identified 14 additional significant quantitative trait loci (QTL), encompassed a total of 42 genes, and indicated that increased marker density contributes to improved statistical power. By integrating gene expression profiling, Gene Ontology (GO) enrichment and haplotype analysis, several potential target genes that may play a role in regulating starch content in maize kernels have been identified. Notably, we found that <i>ZmAPC4</i>, associated with the significant SNP chr4.S_175584318, which encodes a WD40 repeat-like superfamily protein and is highly expressed in maize endosperm, might be a crucial regulator of maize kernel starch synthesis. Out of the 261 inbred lines analyzed, they were categorized into four haplotypes. Remarkably, it was observed that the inbred lines harboring hap4 demonstrated the highest starch content compared to the other haplotypes. Additionally, as a significant achievement, we have developed molecular markers that effectively differentiate maize inbred lines based on their starch content. Overall, our study provides valuable insights into the genetic basis of starch content and the molecular markers can be useful in breeding programs aimed at developing maize varieties with high starch content, thereby improving breeding efficiency.</p>\",\"PeriodicalId\":18769,\"journal\":{\"name\":\"Molecular Breeding\",\"volume\":\"19 1\",\"pages\":\"\"},\"PeriodicalIF\":3.0000,\"publicationDate\":\"2023-12-12\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Molecular Breeding\",\"FirstCategoryId\":\"97\",\"ListUrlMain\":\"https://doi.org/10.1007/s11032-023-01437-6\",\"RegionNum\":3,\"RegionCategory\":\"农林科学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"AGRONOMY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Molecular Breeding","FirstCategoryId":"97","ListUrlMain":"https://doi.org/10.1007/s11032-023-01437-6","RegionNum":3,"RegionCategory":"农林科学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"AGRONOMY","Score":null,"Total":0}
Identification of novel loci associated with starch content in maize kernels by a genome-wide association study using an enlarged SNP panel
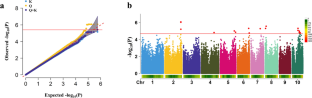
Starch is a major component of cereals, comprising over 70% of dry weight. It serves as a primary carbon source for humans and animals. In addition, starch is an indispensable industrial raw material. While maize (Zea mays) is a key crop and the primary source of starch, the genetic basis for starch content in maize kernels remains poorly understood. In this study, using an enlarged panel, we conducted a genome-wide association study (GWAS) based on best linear unbiased prediction (BLUP) value for starch content of 261 inbred lines across three environments. Compared with previous study, we identified 14 additional significant quantitative trait loci (QTL), encompassed a total of 42 genes, and indicated that increased marker density contributes to improved statistical power. By integrating gene expression profiling, Gene Ontology (GO) enrichment and haplotype analysis, several potential target genes that may play a role in regulating starch content in maize kernels have been identified. Notably, we found that ZmAPC4, associated with the significant SNP chr4.S_175584318, which encodes a WD40 repeat-like superfamily protein and is highly expressed in maize endosperm, might be a crucial regulator of maize kernel starch synthesis. Out of the 261 inbred lines analyzed, they were categorized into four haplotypes. Remarkably, it was observed that the inbred lines harboring hap4 demonstrated the highest starch content compared to the other haplotypes. Additionally, as a significant achievement, we have developed molecular markers that effectively differentiate maize inbred lines based on their starch content. Overall, our study provides valuable insights into the genetic basis of starch content and the molecular markers can be useful in breeding programs aimed at developing maize varieties with high starch content, thereby improving breeding efficiency.
期刊介绍:
Molecular Breeding is an international journal publishing papers on applications of plant molecular biology, i.e., research most likely leading to practical applications. The practical applications might relate to the Developing as well as the industrialised World and have demonstrable benefits for the seed industry, farmers, processing industry, the environment and the consumer.
All papers published should contribute to the understanding and progress of modern plant breeding, encompassing the scientific disciplines of molecular biology, biochemistry, genetics, physiology, pathology, plant breeding, and ecology among others.
Molecular Breeding welcomes the following categories of papers: full papers, short communications, papers describing novel methods and review papers. All submission will be subject to peer review ensuring the highest possible scientific quality standards.
Molecular Breeding core areas:
Molecular Breeding will consider manuscripts describing contemporary methods of molecular genetics and genomic analysis, structural and functional genomics in crops, proteomics and metabolic profiling, abiotic stress and field evaluation of transgenic crops containing particular traits. Manuscripts on marker assisted breeding are also of major interest, in particular novel approaches and new results of marker assisted breeding, QTL cloning, integration of conventional and marker assisted breeding, and QTL studies in crop plants.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
