Nadia Sebbar, Henning Bockhorn, Dimosthenis Trimis
{"title":"1-naphthyl 自由基 C10H7- 与氧气的氧化反应:热化学、动力学和可能的反应途径","authors":"Nadia Sebbar, Henning Bockhorn, Dimosthenis Trimis","doi":"10.1002/kin.21702","DOIUrl":null,"url":null,"abstract":"<p>The reaction of the 1-naphthyl radical C<sub>10</sub>H<sub>7</sub>• (A2•) with molecular (<sup>3</sup>O<sub>2</sub>) and atomic oxygen, as part of the oxidation reactions of naphthalene, is examined using ab-initio and DFT quantum chemistry calculations. The study focuses on pathways that produce the intermediate final products CO, phenyl and C<sub>2</sub>H<sub>2</sub>, which may constitute a repetitive reaction sequence for the successive diminution of six-membered rings also in larger polycyclic aromatic hydrocarbons. The primary attack of <sup>3</sup>O<sub>2</sub> on the 1-naphthyl radical leads to a peroxy radical C<sub>10</sub>H<sub>7</sub>OO• (A2OO•), which undergoes further propagation and/or chain branching reactions. The thermochemistry of intermediates and transition state structures is investigated as well as the identification of all plausible reaction pathways for the A2• + O<sub>2</sub> / A2• + O systems. Structures and enthalpies of formation for the involved species are reported along with transition state barriers and reaction pathways. Standard enthalpies of formation are calculated using ab initio (CBS-QB3) and DFT calculations (B3LYP, M06, APFD). The reaction of A2• with <sup>3</sup>O<sub>2</sub> opens six main consecutive reaction channels with new ones not currently considered in oxidation mechanisms. The reaction paths comprise important exothermic chain branching reactions and the formation of unsaturated oxygenated hydrocarbon intermediates. The primary attack of <sup>3</sup>O<sub>2</sub> at the A2• radical has a well depth of some 50 kcal mol<sup>−1</sup> while the six consecutive channels exhibit energy barriers below the energy of the A2• radical. The kinetic parameters of each path are determined using chemical activation analysis based on the canonical transition state theory calculations. The investigated reactions could serve as part of a comprehensive mechanism for the oxidation of naphthalene. The principal result from this study is that the consecutive reactions of the A2• radical, viz. the channels conducting to a phenyl radical C<sub>6</sub>H<sub>5</sub>•, CO<sub>2</sub>, CO (which oxidized to CO<sub>2</sub>) and C<sub>2</sub>H<sub>2</sub> are by orders of magnitude faster than the activation of naphthalene by oxygen (A2 + O<sub>2</sub> → A2• + HO<sub>2</sub>).</p>","PeriodicalId":13894,"journal":{"name":"International Journal of Chemical Kinetics","volume":"56 4","pages":"210-232"},"PeriodicalIF":1.6000,"publicationDate":"2023-12-12","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1002/kin.21702","citationCount":"0","resultStr":"{\"title\":\"Oxidation of the 1-naphthyl radical C10H7• with oxygen: Thermochemistry, kinetics, and possible reaction pathways\",\"authors\":\"Nadia Sebbar, Henning Bockhorn, Dimosthenis Trimis\",\"doi\":\"10.1002/kin.21702\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p>The reaction of the 1-naphthyl radical C<sub>10</sub>H<sub>7</sub>• (A2•) with molecular (<sup>3</sup>O<sub>2</sub>) and atomic oxygen, as part of the oxidation reactions of naphthalene, is examined using ab-initio and DFT quantum chemistry calculations. The study focuses on pathways that produce the intermediate final products CO, phenyl and C<sub>2</sub>H<sub>2</sub>, which may constitute a repetitive reaction sequence for the successive diminution of six-membered rings also in larger polycyclic aromatic hydrocarbons. The primary attack of <sup>3</sup>O<sub>2</sub> on the 1-naphthyl radical leads to a peroxy radical C<sub>10</sub>H<sub>7</sub>OO• (A2OO•), which undergoes further propagation and/or chain branching reactions. The thermochemistry of intermediates and transition state structures is investigated as well as the identification of all plausible reaction pathways for the A2• + O<sub>2</sub> / A2• + O systems. Structures and enthalpies of formation for the involved species are reported along with transition state barriers and reaction pathways. Standard enthalpies of formation are calculated using ab initio (CBS-QB3) and DFT calculations (B3LYP, M06, APFD). The reaction of A2• with <sup>3</sup>O<sub>2</sub> opens six main consecutive reaction channels with new ones not currently considered in oxidation mechanisms. The reaction paths comprise important exothermic chain branching reactions and the formation of unsaturated oxygenated hydrocarbon intermediates. The primary attack of <sup>3</sup>O<sub>2</sub> at the A2• radical has a well depth of some 50 kcal mol<sup>−1</sup> while the six consecutive channels exhibit energy barriers below the energy of the A2• radical. The kinetic parameters of each path are determined using chemical activation analysis based on the canonical transition state theory calculations. The investigated reactions could serve as part of a comprehensive mechanism for the oxidation of naphthalene. The principal result from this study is that the consecutive reactions of the A2• radical, viz. the channels conducting to a phenyl radical C<sub>6</sub>H<sub>5</sub>•, CO<sub>2</sub>, CO (which oxidized to CO<sub>2</sub>) and C<sub>2</sub>H<sub>2</sub> are by orders of magnitude faster than the activation of naphthalene by oxygen (A2 + O<sub>2</sub> → A2• + HO<sub>2</sub>).</p>\",\"PeriodicalId\":13894,\"journal\":{\"name\":\"International Journal of Chemical Kinetics\",\"volume\":\"56 4\",\"pages\":\"210-232\"},\"PeriodicalIF\":1.6000,\"publicationDate\":\"2023-12-12\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://onlinelibrary.wiley.com/doi/epdf/10.1002/kin.21702\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"International Journal of Chemical Kinetics\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://onlinelibrary.wiley.com/doi/10.1002/kin.21702\",\"RegionNum\":4,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q4\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"International Journal of Chemical Kinetics","FirstCategoryId":"92","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/kin.21702","RegionNum":4,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q4","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
Oxidation of the 1-naphthyl radical C10H7• with oxygen: Thermochemistry, kinetics, and possible reaction pathways
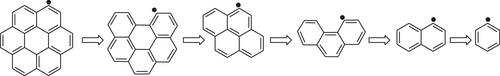
The reaction of the 1-naphthyl radical C10H7• (A2•) with molecular (3O2) and atomic oxygen, as part of the oxidation reactions of naphthalene, is examined using ab-initio and DFT quantum chemistry calculations. The study focuses on pathways that produce the intermediate final products CO, phenyl and C2H2, which may constitute a repetitive reaction sequence for the successive diminution of six-membered rings also in larger polycyclic aromatic hydrocarbons. The primary attack of 3O2 on the 1-naphthyl radical leads to a peroxy radical C10H7OO• (A2OO•), which undergoes further propagation and/or chain branching reactions. The thermochemistry of intermediates and transition state structures is investigated as well as the identification of all plausible reaction pathways for the A2• + O2 / A2• + O systems. Structures and enthalpies of formation for the involved species are reported along with transition state barriers and reaction pathways. Standard enthalpies of formation are calculated using ab initio (CBS-QB3) and DFT calculations (B3LYP, M06, APFD). The reaction of A2• with 3O2 opens six main consecutive reaction channels with new ones not currently considered in oxidation mechanisms. The reaction paths comprise important exothermic chain branching reactions and the formation of unsaturated oxygenated hydrocarbon intermediates. The primary attack of 3O2 at the A2• radical has a well depth of some 50 kcal mol−1 while the six consecutive channels exhibit energy barriers below the energy of the A2• radical. The kinetic parameters of each path are determined using chemical activation analysis based on the canonical transition state theory calculations. The investigated reactions could serve as part of a comprehensive mechanism for the oxidation of naphthalene. The principal result from this study is that the consecutive reactions of the A2• radical, viz. the channels conducting to a phenyl radical C6H5•, CO2, CO (which oxidized to CO2) and C2H2 are by orders of magnitude faster than the activation of naphthalene by oxygen (A2 + O2 → A2• + HO2).
期刊介绍:
As the leading archival journal devoted exclusively to chemical kinetics, the International Journal of Chemical Kinetics publishes original research in gas phase, condensed phase, and polymer reaction kinetics, as well as biochemical and surface kinetics. The Journal seeks to be the primary archive for careful experimental measurements of reaction kinetics, in both simple and complex systems. The Journal also presents new developments in applied theoretical kinetics and publishes large kinetic models, and the algorithms and estimates used in these models. These include methods for handling the large reaction networks important in biochemistry, catalysis, and free radical chemistry. In addition, the Journal explores such topics as the quantitative relationships between molecular structure and chemical reactivity, organic/inorganic chemistry and reaction mechanisms, and the reactive chemistry at interfaces.


 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
