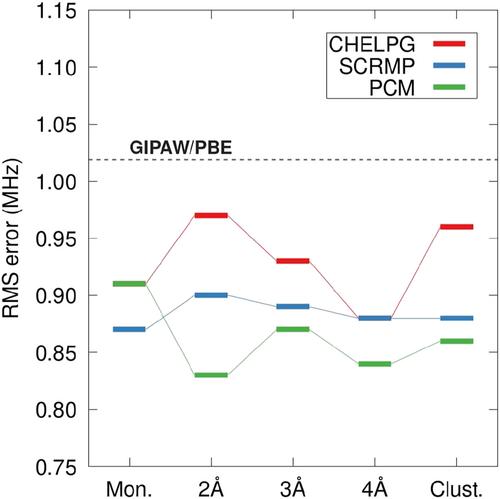
{"title":"预测分子晶体中的 51 V 核磁共振观测值。","authors":"Joshua D. Hartman, Daniel Capistran","doi":"10.1002/mrc.5420","DOIUrl":null,"url":null,"abstract":"<p>Solid-state nuclear magnetic resonance (NMR) spectroscopy and quantum chemical density functional theory (DFT) calculations are widely used to characterize vanadium centers in biological and pharmaceutically relevant compounds. Several techniques have been recently developed to improve the accuracy of predicted NMR parameters obtained from DFT. Fragment-based and planewave-corrected methods employing hybrid density functionals are particularly effective tools for solid-state applications. A recent benchmark study involving molecular crystal compounds found that fragment-based NMR calculations using hybrid density functionals improve the accuracy of predicted <sup>51</sup>V chemical shieldings by 20% relative to traditional planewave methods. This work extends the previous study, including a careful analysis of <sup>51</sup>V chemical shift anisotropy, electric field gradient calculations, and a more extensive test set. The accuracy of planewave-corrected techniques and recently developed fragment-based methods using electrostatic embedding based on the polarized continuum model (PCM) are found to be highly competitive with previous methods. Planewave-corrected methods achieve a 34% improvement in the errors of predicted <sup>51</sup>V chemical shieldings relative to planewave. Additionally, planewave-corrected and fragment-based calculations were performed using PCM embedding, improving the accuracy of predicted <sup>51</sup>V chemical shielding (CS) tensor principal values by 30% and \n<span></span><math>\n <msub>\n <mrow>\n <mi>C</mi>\n </mrow>\n <mrow>\n <mi>q</mi>\n </mrow>\n </msub></math> values by 15% relative to traditional planewave methods. The performance of these methods is further examined using a redox-active oxovandium complex and a common <sup>51</sup>V NMR reference compound.</p>","PeriodicalId":18142,"journal":{"name":"Magnetic Resonance in Chemistry","volume":"62 6","pages":"416-428"},"PeriodicalIF":1.4000,"publicationDate":"2023-12-19","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1002/mrc.5420","citationCount":"0","resultStr":"{\"title\":\"Predicting 51V nuclear magnetic resonance observables in molecular crystals\",\"authors\":\"Joshua D. Hartman, Daniel Capistran\",\"doi\":\"10.1002/mrc.5420\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p>Solid-state nuclear magnetic resonance (NMR) spectroscopy and quantum chemical density functional theory (DFT) calculations are widely used to characterize vanadium centers in biological and pharmaceutically relevant compounds. Several techniques have been recently developed to improve the accuracy of predicted NMR parameters obtained from DFT. Fragment-based and planewave-corrected methods employing hybrid density functionals are particularly effective tools for solid-state applications. A recent benchmark study involving molecular crystal compounds found that fragment-based NMR calculations using hybrid density functionals improve the accuracy of predicted <sup>51</sup>V chemical shieldings by 20% relative to traditional planewave methods. This work extends the previous study, including a careful analysis of <sup>51</sup>V chemical shift anisotropy, electric field gradient calculations, and a more extensive test set. The accuracy of planewave-corrected techniques and recently developed fragment-based methods using electrostatic embedding based on the polarized continuum model (PCM) are found to be highly competitive with previous methods. Planewave-corrected methods achieve a 34% improvement in the errors of predicted <sup>51</sup>V chemical shieldings relative to planewave. Additionally, planewave-corrected and fragment-based calculations were performed using PCM embedding, improving the accuracy of predicted <sup>51</sup>V chemical shielding (CS) tensor principal values by 30% and \\n<span></span><math>\\n <msub>\\n <mrow>\\n <mi>C</mi>\\n </mrow>\\n <mrow>\\n <mi>q</mi>\\n </mrow>\\n </msub></math> values by 15% relative to traditional planewave methods. The performance of these methods is further examined using a redox-active oxovandium complex and a common <sup>51</sup>V NMR reference compound.</p>\",\"PeriodicalId\":18142,\"journal\":{\"name\":\"Magnetic Resonance in Chemistry\",\"volume\":\"62 6\",\"pages\":\"416-428\"},\"PeriodicalIF\":1.4000,\"publicationDate\":\"2023-12-19\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://onlinelibrary.wiley.com/doi/epdf/10.1002/mrc.5420\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Magnetic Resonance in Chemistry\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://analyticalsciencejournals.onlinelibrary.wiley.com/doi/10.1002/mrc.5420\",\"RegionNum\":3,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q3\",\"JCRName\":\"CHEMISTRY, MULTIDISCIPLINARY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Magnetic Resonance in Chemistry","FirstCategoryId":"92","ListUrlMain":"https://analyticalsciencejournals.onlinelibrary.wiley.com/doi/10.1002/mrc.5420","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"CHEMISTRY, MULTIDISCIPLINARY","Score":null,"Total":0}
引用次数: 0
摘要
固态核磁共振(NMR)光谱和量子化学密度泛函理论(DFT)计算被广泛用于表征生物和制药相关化合物中的钒中心。最近开发了几种技术,以提高从 DFT 中获得的 NMR 参数预测的准确性。采用混合密度函数的基于片段和平面波校正的方法是固态应用中特别有效的工具。最近一项涉及分子晶体化合物的基准研究发现,与传统的平面波方法相比,采用混合密度函数的基于片段的 NMR 计算可将 51 V 化学屏蔽的预测精度提高 20%。这项工作扩展了之前的研究,包括对 51 V 化学位移各向异性的仔细分析、电场梯度计算和更广泛的测试集。结果发现,平面波校正技术和最近开发的基于极化连续体模型(PCM)的静电嵌入片段法的准确性与以前的方法相比具有很强的竞争力。相对于平面波,平面波校正方法将 51 V 化学屏蔽的预测误差提高了 34%。此外,使用 PCM 嵌入进行了平面波校正和基于片段的计算,与传统的平面波方法相比,51 V 化学屏蔽 (CS) 张量主值的预测精度提高了 30%,C q $$ {C}_q $$ 值的预测精度提高了 15%。使用氧化还原活性氧钒复合物和常见的 51 V NMR 参考化合物进一步检验了这些方法的性能。
Predicting 51V nuclear magnetic resonance observables in molecular crystals
Solid-state nuclear magnetic resonance (NMR) spectroscopy and quantum chemical density functional theory (DFT) calculations are widely used to characterize vanadium centers in biological and pharmaceutically relevant compounds. Several techniques have been recently developed to improve the accuracy of predicted NMR parameters obtained from DFT. Fragment-based and planewave-corrected methods employing hybrid density functionals are particularly effective tools for solid-state applications. A recent benchmark study involving molecular crystal compounds found that fragment-based NMR calculations using hybrid density functionals improve the accuracy of predicted 51V chemical shieldings by 20% relative to traditional planewave methods. This work extends the previous study, including a careful analysis of 51V chemical shift anisotropy, electric field gradient calculations, and a more extensive test set. The accuracy of planewave-corrected techniques and recently developed fragment-based methods using electrostatic embedding based on the polarized continuum model (PCM) are found to be highly competitive with previous methods. Planewave-corrected methods achieve a 34% improvement in the errors of predicted 51V chemical shieldings relative to planewave. Additionally, planewave-corrected and fragment-based calculations were performed using PCM embedding, improving the accuracy of predicted 51V chemical shielding (CS) tensor principal values by 30% and
values by 15% relative to traditional planewave methods. The performance of these methods is further examined using a redox-active oxovandium complex and a common 51V NMR reference compound.
期刊介绍:
MRC is devoted to the rapid publication of papers which are concerned with the development of magnetic resonance techniques, or in which the application of such techniques plays a pivotal part. Contributions from scientists working in all areas of NMR, ESR and NQR are invited, and papers describing applications in all branches of chemistry, structural biology and materials chemistry are published.
The journal is of particular interest not only to scientists working in academic research, but also those working in commercial organisations who need to keep up-to-date with the latest practical applications of magnetic resonance techniques.





 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
