{"title":"早发慢性角膜炎是自身免疫性多内分泌病综合征 1 型(APS-1)的首发症状:病例报告与文献综述。","authors":"Enver Şimşek, Tülay Şimşek, Oğuz Çilingir","doi":"10.4274/jcrpe.galenos.2023.2023-9-17","DOIUrl":null,"url":null,"abstract":"<p><p>Autoimmune polyendocrine syndrome type 1 (APS-1), also referred to as autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy, is a rare monogenic autosomal recessive autoimmune disease. It is caused by mutations in the autoimmune regulator (<i>AIRE</i>) gene. APS-1 is diagnosed clinically by the presence of two of the three major components: chronic mucocutaneous candidiasis, hypoparathyroidism (HPT), and primary adrenocortical insufficiency. A 3.3-year-old girl presented with a carpopedal spasm to the pediatric emergency clinic. She had a history of recurrent keratitis, and chronic candidiasis, manifesting as urinary tract infections and oral thrush. HPT was diagnosed based on low serum concentrations of calcium and parathyroid hormone and elevated serum concentrations of phosphate, and treatment with calcium and calcitriol supplementation was started. Genetic testing identified a homozygous nonsense mutation, c.769C>T (p.R257X), in exon 6 of <i>AIRE</i> which has been reported previously. At the age of 5.6 years, she presented with adrenal crisis, and treatment with hydrocortisone and fludrocortisone was started. This case demonstrates that unexplained chronic keratitis in children may be the first and most severe component of this syndrome. The classic triad of APS-1 may also appear in the first decade of life.</p>","PeriodicalId":48805,"journal":{"name":"Journal of Clinical Research in Pediatric Endocrinology","volume":" ","pages":"359-364"},"PeriodicalIF":1.5000,"publicationDate":"2025-08-22","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12372638/pdf/","citationCount":"0","resultStr":"{\"title\":\"Early-onset Chronic Keratitis as the First Presenting Component of Autoimmune Polyendocrine Syndrome Type 1: A Case Report and Review of the Literature\",\"authors\":\"Enver Şimşek, Tülay Şimşek, Oğuz Çilingir\",\"doi\":\"10.4274/jcrpe.galenos.2023.2023-9-17\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Autoimmune polyendocrine syndrome type 1 (APS-1), also referred to as autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy, is a rare monogenic autosomal recessive autoimmune disease. It is caused by mutations in the autoimmune regulator (<i>AIRE</i>) gene. APS-1 is diagnosed clinically by the presence of two of the three major components: chronic mucocutaneous candidiasis, hypoparathyroidism (HPT), and primary adrenocortical insufficiency. A 3.3-year-old girl presented with a carpopedal spasm to the pediatric emergency clinic. She had a history of recurrent keratitis, and chronic candidiasis, manifesting as urinary tract infections and oral thrush. HPT was diagnosed based on low serum concentrations of calcium and parathyroid hormone and elevated serum concentrations of phosphate, and treatment with calcium and calcitriol supplementation was started. Genetic testing identified a homozygous nonsense mutation, c.769C>T (p.R257X), in exon 6 of <i>AIRE</i> which has been reported previously. At the age of 5.6 years, she presented with adrenal crisis, and treatment with hydrocortisone and fludrocortisone was started. This case demonstrates that unexplained chronic keratitis in children may be the first and most severe component of this syndrome. The classic triad of APS-1 may also appear in the first decade of life.</p>\",\"PeriodicalId\":48805,\"journal\":{\"name\":\"Journal of Clinical Research in Pediatric Endocrinology\",\"volume\":\" \",\"pages\":\"359-364\"},\"PeriodicalIF\":1.5000,\"publicationDate\":\"2025-08-22\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12372638/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Clinical Research in Pediatric Endocrinology\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://doi.org/10.4274/jcrpe.galenos.2023.2023-9-17\",\"RegionNum\":4,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2023/12/12 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"Q4\",\"JCRName\":\"ENDOCRINOLOGY & METABOLISM\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Clinical Research in Pediatric Endocrinology","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.4274/jcrpe.galenos.2023.2023-9-17","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2023/12/12 0:00:00","PubModel":"Epub","JCR":"Q4","JCRName":"ENDOCRINOLOGY & METABOLISM","Score":null,"Total":0}
Early-onset Chronic Keratitis as the First Presenting Component of Autoimmune Polyendocrine Syndrome Type 1: A Case Report and Review of the Literature
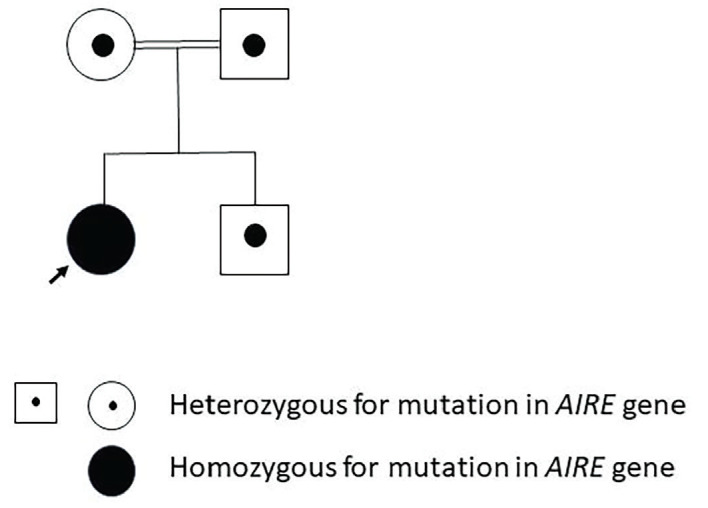
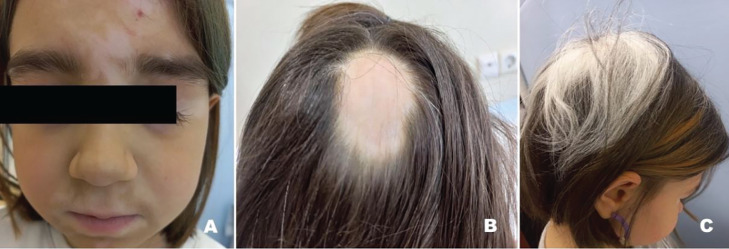
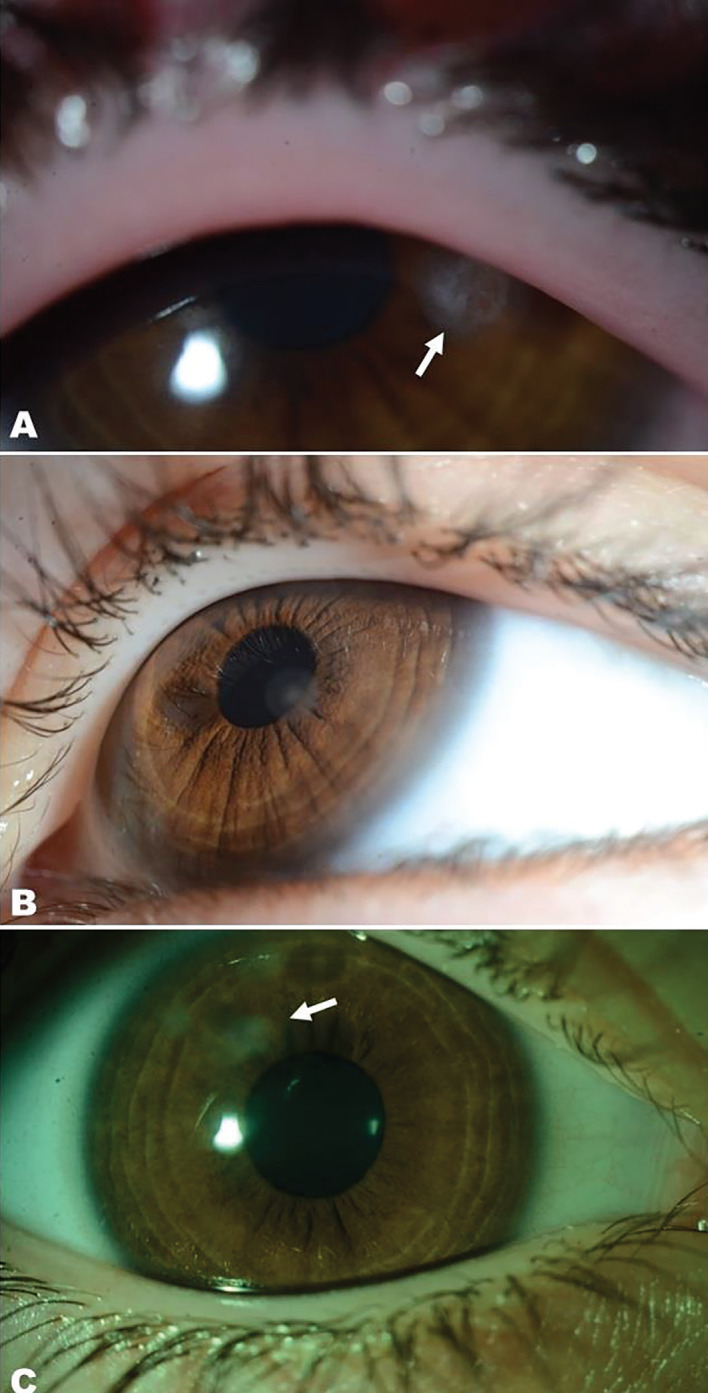
Autoimmune polyendocrine syndrome type 1 (APS-1), also referred to as autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy, is a rare monogenic autosomal recessive autoimmune disease. It is caused by mutations in the autoimmune regulator (AIRE) gene. APS-1 is diagnosed clinically by the presence of two of the three major components: chronic mucocutaneous candidiasis, hypoparathyroidism (HPT), and primary adrenocortical insufficiency. A 3.3-year-old girl presented with a carpopedal spasm to the pediatric emergency clinic. She had a history of recurrent keratitis, and chronic candidiasis, manifesting as urinary tract infections and oral thrush. HPT was diagnosed based on low serum concentrations of calcium and parathyroid hormone and elevated serum concentrations of phosphate, and treatment with calcium and calcitriol supplementation was started. Genetic testing identified a homozygous nonsense mutation, c.769C>T (p.R257X), in exon 6 of AIRE which has been reported previously. At the age of 5.6 years, she presented with adrenal crisis, and treatment with hydrocortisone and fludrocortisone was started. This case demonstrates that unexplained chronic keratitis in children may be the first and most severe component of this syndrome. The classic triad of APS-1 may also appear in the first decade of life.
期刊介绍:
The Journal of Clinical Research in Pediatric Endocrinology (JCRPE) publishes original research articles, reviews, short communications, letters, case reports and other special features related to the field of pediatric endocrinology. JCRPE is published in English by the Turkish Pediatric Endocrinology and Diabetes Society quarterly (March, June, September, December). The target audience is physicians, researchers and other healthcare professionals in all areas of pediatric endocrinology.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
