{"title":"特发性孤立性促肾上腺皮质激素缺乏症:对一种异质性和报告不足的疾病的系统回顾。","authors":"E Van Mieghem, C De Block, C De Herdt","doi":"10.1007/s11102-023-01366-9","DOIUrl":null,"url":null,"abstract":"<p><p>Isolated adrenocorticotropic hormone deficiency (IAD) is considered to be a rare disease. Due to the nonspecific clinical presentation, precise data on the prevalence and incidence are lacking. In this systematic review, we aimed to analyse the clinical characteristics, association with autoimmune diseases, and management of acquired idiopathic IAD cases. A structured search was conducted after developing a search strategy combining terms for acquired (idiopathic) IAD. Articles describing an adult case with a diagnosis of ACTH deficiency using dynamic testing, no deficiency of other pituitary axes, and MRI of the brain/pituitary protocolled as normal, were included. Exclusion criteria were cases describing congenital IAD, cases with another aetiology for IAD, and articles where full text was not available. In total 42 articles were included, consisting of 85 cases of acquired idiopathic IAD. Distribution by sex was approximately equal (F:M; 47:38). Lethargy was the most common presenting symptom (38%), followed by weight loss (25%), anorexia (22%), and myalgia/arthralgia (12%). Eight cases (9.5%) presented with an Addison crisis. 31% of cases had an autoimmune disease at diagnosis of which Hashimoto hypothyroidism was the most frequent. Data about follow-up was scarce; dynamic testing was repeated in 4 cases of which 2 showed recovery of the adrenal axis. We report the largest case series of acquired idiopathic IAD to date. Our systematic review highlights the lack of a clear definition and diagnostic work-up. Based on the findings in this review a proposition is made for a flowchart to diagnose acquired idiopathic IAD.</p>","PeriodicalId":20202,"journal":{"name":"Pituitary","volume":" ","pages":"23-32"},"PeriodicalIF":3.4000,"publicationDate":"2024-02-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Idiopathic isolated adrenocorticotropic hormone deficiency: a systematic review of a heterogeneous and underreported disease.\",\"authors\":\"E Van Mieghem, C De Block, C De Herdt\",\"doi\":\"10.1007/s11102-023-01366-9\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Isolated adrenocorticotropic hormone deficiency (IAD) is considered to be a rare disease. Due to the nonspecific clinical presentation, precise data on the prevalence and incidence are lacking. In this systematic review, we aimed to analyse the clinical characteristics, association with autoimmune diseases, and management of acquired idiopathic IAD cases. A structured search was conducted after developing a search strategy combining terms for acquired (idiopathic) IAD. Articles describing an adult case with a diagnosis of ACTH deficiency using dynamic testing, no deficiency of other pituitary axes, and MRI of the brain/pituitary protocolled as normal, were included. Exclusion criteria were cases describing congenital IAD, cases with another aetiology for IAD, and articles where full text was not available. In total 42 articles were included, consisting of 85 cases of acquired idiopathic IAD. Distribution by sex was approximately equal (F:M; 47:38). Lethargy was the most common presenting symptom (38%), followed by weight loss (25%), anorexia (22%), and myalgia/arthralgia (12%). Eight cases (9.5%) presented with an Addison crisis. 31% of cases had an autoimmune disease at diagnosis of which Hashimoto hypothyroidism was the most frequent. Data about follow-up was scarce; dynamic testing was repeated in 4 cases of which 2 showed recovery of the adrenal axis. We report the largest case series of acquired idiopathic IAD to date. Our systematic review highlights the lack of a clear definition and diagnostic work-up. Based on the findings in this review a proposition is made for a flowchart to diagnose acquired idiopathic IAD.</p>\",\"PeriodicalId\":20202,\"journal\":{\"name\":\"Pituitary\",\"volume\":\" \",\"pages\":\"23-32\"},\"PeriodicalIF\":3.4000,\"publicationDate\":\"2024-02-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Pituitary\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://doi.org/10.1007/s11102-023-01366-9\",\"RegionNum\":2,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2023/12/27 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"Q2\",\"JCRName\":\"ENDOCRINOLOGY & METABOLISM\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Pituitary","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1007/s11102-023-01366-9","RegionNum":2,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2023/12/27 0:00:00","PubModel":"Epub","JCR":"Q2","JCRName":"ENDOCRINOLOGY & METABOLISM","Score":null,"Total":0}
引用次数: 0
摘要
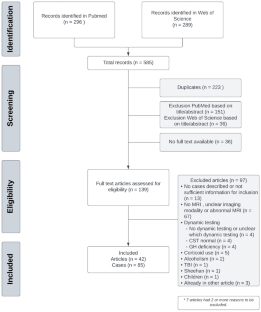
孤立性促肾上腺皮质激素缺乏症(IAD)被认为是一种罕见疾病。由于其临床表现无特异性,目前尚缺乏有关患病率和发病率的精确数据。在这篇系统性综述中,我们旨在分析获得性特发性 IAD 病例的临床特征、与自身免疫性疾病的关联以及治疗方法。在制定了结合获得性(特发性)IAD术语的检索策略后,我们进行了结构化检索。其中包括描述成人病例的文章,这些病例通过动态检测确诊为促肾上腺皮质激素(ACTH)缺乏症,没有其他垂体轴的缺乏症,脑/垂体核磁共振成像显示正常。排除标准包括描述先天性 IAD 的病例、IAD 有其他病因的病例以及无法获得全文的文章。共纳入 42 篇文章,包括 85 例获得性特发性 IAD 病例。性别分布大致相同(女:男;47:38)。嗜睡是最常见的首发症状(38%),其次是体重减轻(25%)、厌食(22%)和肌痛/关节痛(12%)。8例病例(9.5%)出现阿狄森危象。31%的病例在确诊时患有自身免疫性疾病,其中桥本甲状腺功能减退症最为常见。有关随访的数据很少;有4例病例重复进行了动态检测,其中2例显示肾上腺轴功能恢复。我们报告了迄今为止最大的获得性特发性 IAD 病例系列。我们的系统回顾强调了缺乏明确定义和诊断方法的问题。根据本综述的研究结果,我们提出了诊断获得性特发性 IAD 的流程图。
Idiopathic isolated adrenocorticotropic hormone deficiency: a systematic review of a heterogeneous and underreported disease.
Isolated adrenocorticotropic hormone deficiency (IAD) is considered to be a rare disease. Due to the nonspecific clinical presentation, precise data on the prevalence and incidence are lacking. In this systematic review, we aimed to analyse the clinical characteristics, association with autoimmune diseases, and management of acquired idiopathic IAD cases. A structured search was conducted after developing a search strategy combining terms for acquired (idiopathic) IAD. Articles describing an adult case with a diagnosis of ACTH deficiency using dynamic testing, no deficiency of other pituitary axes, and MRI of the brain/pituitary protocolled as normal, were included. Exclusion criteria were cases describing congenital IAD, cases with another aetiology for IAD, and articles where full text was not available. In total 42 articles were included, consisting of 85 cases of acquired idiopathic IAD. Distribution by sex was approximately equal (F:M; 47:38). Lethargy was the most common presenting symptom (38%), followed by weight loss (25%), anorexia (22%), and myalgia/arthralgia (12%). Eight cases (9.5%) presented with an Addison crisis. 31% of cases had an autoimmune disease at diagnosis of which Hashimoto hypothyroidism was the most frequent. Data about follow-up was scarce; dynamic testing was repeated in 4 cases of which 2 showed recovery of the adrenal axis. We report the largest case series of acquired idiopathic IAD to date. Our systematic review highlights the lack of a clear definition and diagnostic work-up. Based on the findings in this review a proposition is made for a flowchart to diagnose acquired idiopathic IAD.
期刊介绍:
Pituitary is an international publication devoted to basic and clinical aspects of the pituitary gland. It is designed to publish original, high quality research in both basic and pituitary function as well as clinical pituitary disease.
The journal considers:
Biology of Pituitary Tumors
Mechanisms of Pituitary Hormone Secretion
Regulation of Pituitary Function
Prospective Clinical Studies of Pituitary Disease
Critical Basic and Clinical Reviews
Pituitary is directed at basic investigators, physiologists, clinical adult and pediatric endocrinologists, neurosurgeons and reproductive endocrinologists interested in the broad field of the pituitary and its disorders. The Editorial Board has been drawn from international experts in basic and clinical endocrinology. The journal offers a rapid turnaround time for review of manuscripts, and the high standard of the journal is maintained by a selective peer-review process which aims to publish only the highest quality manuscripts. Pituitary will foster the publication of creative scholarship as it pertains to the pituitary and will provide a forum for basic scientists and clinicians to publish their high quality pituitary-related work.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
