Kritika Krishnamurthy, Jui Choudhuri, K H Ramesh, Yanhua Wang
{"title":"MPO阴性急性早幼粒细胞白血病中背景中性粒细胞的MPO表达,是确诊难题的简单线索:病例报告与文献综述。","authors":"Kritika Krishnamurthy, Jui Choudhuri, K H Ramesh, Yanhua Wang","doi":"10.1155/2023/7979261","DOIUrl":null,"url":null,"abstract":"<p><p>Acute promyelocytic leukemia (APL) is characterized by the pathogenic driver fusion transcript PML-RARA resulting from the t(15;17) translocation. Early recognition of APL with prompt ATRA induction has a decisive impact on the early death rate. The preliminary diagnosis of APL relies heavily on cytomorphology and flow cytometry. In APL with variant morphology, such as the microgranular variant, immunophenotype, especially the bright MPO positivity is the basis of diagnosis. Till date, only five cases of APL with reduced/absent MPO have been described in literature. The identification of MPO deficiency based on genetic testing would involve at the least a MPO gene scanning with NGS, followed by microarray to identify somatic uniparental disomy in heterozygotes. This testing is not only redundant given the scant clinical implications of heterozygous MPO deficiency but also time consuming. An easy way to identify background MPO deficiency confounding the immunophenotype of a myeloid neoplasm is the MPO expression in background neutrophils gated on the initial flow cytometry. A dim MPO in the background neutrophils, in the morphological setting of APL, can identify underlying MPO deficiency, clarifying the immunophenotypic ambiguity and thus establishing an unequivocal diagnosis as seen in the current case.</p>","PeriodicalId":46307,"journal":{"name":"Case Reports in Hematology","volume":"2023 ","pages":"7979261"},"PeriodicalIF":0.7000,"publicationDate":"2023-12-26","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10761215/pdf/","citationCount":"0","resultStr":"{\"title\":\"MPO Expression of Background Neutrophils in MPO Negative Acute Promyelocytic Leukemia, An Easy Clue to Corroborate a Challenging Diagnosis: A Case Report and Review of Literature.\",\"authors\":\"Kritika Krishnamurthy, Jui Choudhuri, K H Ramesh, Yanhua Wang\",\"doi\":\"10.1155/2023/7979261\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Acute promyelocytic leukemia (APL) is characterized by the pathogenic driver fusion transcript PML-RARA resulting from the t(15;17) translocation. Early recognition of APL with prompt ATRA induction has a decisive impact on the early death rate. The preliminary diagnosis of APL relies heavily on cytomorphology and flow cytometry. In APL with variant morphology, such as the microgranular variant, immunophenotype, especially the bright MPO positivity is the basis of diagnosis. Till date, only five cases of APL with reduced/absent MPO have been described in literature. The identification of MPO deficiency based on genetic testing would involve at the least a MPO gene scanning with NGS, followed by microarray to identify somatic uniparental disomy in heterozygotes. This testing is not only redundant given the scant clinical implications of heterozygous MPO deficiency but also time consuming. An easy way to identify background MPO deficiency confounding the immunophenotype of a myeloid neoplasm is the MPO expression in background neutrophils gated on the initial flow cytometry. A dim MPO in the background neutrophils, in the morphological setting of APL, can identify underlying MPO deficiency, clarifying the immunophenotypic ambiguity and thus establishing an unequivocal diagnosis as seen in the current case.</p>\",\"PeriodicalId\":46307,\"journal\":{\"name\":\"Case Reports in Hematology\",\"volume\":\"2023 \",\"pages\":\"7979261\"},\"PeriodicalIF\":0.7000,\"publicationDate\":\"2023-12-26\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10761215/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Case Reports in Hematology\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1155/2023/7979261\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2023/1/1 0:00:00\",\"PubModel\":\"eCollection\",\"JCR\":\"Q4\",\"JCRName\":\"HEMATOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Case Reports in Hematology","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1155/2023/7979261","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2023/1/1 0:00:00","PubModel":"eCollection","JCR":"Q4","JCRName":"HEMATOLOGY","Score":null,"Total":0}
MPO Expression of Background Neutrophils in MPO Negative Acute Promyelocytic Leukemia, An Easy Clue to Corroborate a Challenging Diagnosis: A Case Report and Review of Literature.
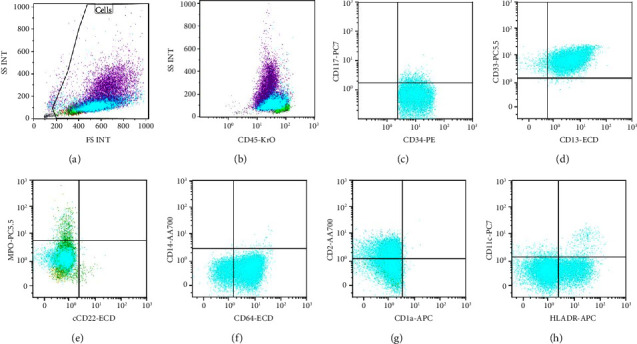
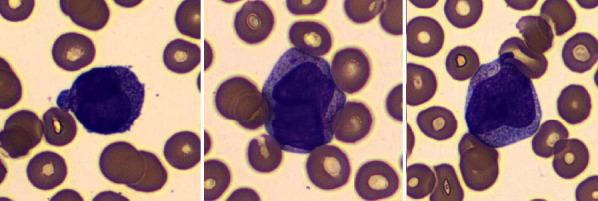
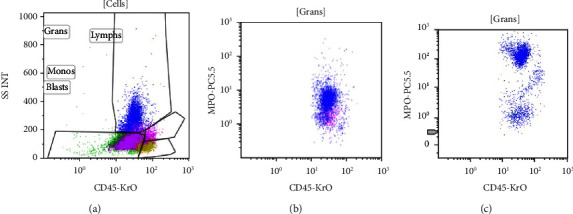
Acute promyelocytic leukemia (APL) is characterized by the pathogenic driver fusion transcript PML-RARA resulting from the t(15;17) translocation. Early recognition of APL with prompt ATRA induction has a decisive impact on the early death rate. The preliminary diagnosis of APL relies heavily on cytomorphology and flow cytometry. In APL with variant morphology, such as the microgranular variant, immunophenotype, especially the bright MPO positivity is the basis of diagnosis. Till date, only five cases of APL with reduced/absent MPO have been described in literature. The identification of MPO deficiency based on genetic testing would involve at the least a MPO gene scanning with NGS, followed by microarray to identify somatic uniparental disomy in heterozygotes. This testing is not only redundant given the scant clinical implications of heterozygous MPO deficiency but also time consuming. An easy way to identify background MPO deficiency confounding the immunophenotype of a myeloid neoplasm is the MPO expression in background neutrophils gated on the initial flow cytometry. A dim MPO in the background neutrophils, in the morphological setting of APL, can identify underlying MPO deficiency, clarifying the immunophenotypic ambiguity and thus establishing an unequivocal diagnosis as seen in the current case.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
