{"title":"先天性肌肉萎缩症","authors":"Haluk Topaloğlu, Bita Poorshiri","doi":"10.1002/cns3.20050","DOIUrl":null,"url":null,"abstract":"<div>\n \n \n <section>\n \n <h3> Background</h3>\n \n <p>Congenital muscular dystrophies (CMDs) are genetically and clinically heterogeneous inherited conditions. Onset is typically within the first year of life. Most CMDs are autosomal recessive, except for de novo dominant mutations in <i>LMNA</i>-related muscular dystrophy and some collagen-6-associated disorders.</p>\n </section>\n \n <section>\n \n <h3> Results</h3>\n \n <p>CMD is characterized by progressive muscular weakness, hypotonia, multiple contractures with a variable degree, spinal stiffness, delay in motor milestones acquisition, and histologically dystrophic lesions. Also, some forms of CMD may feature structural and myelination abnormalities on brain magnetic resonance imaging, intellectual impairment, and structural abnormalities of the eye. Muscle biopsy specimens exhibit a dystrophic pattern, but the appearance is quite variable depending on the different stages and severity of the disorder. The prevalence of CMD is estimated to be one in 100 000 people. Over the last few years, with advances in molecular genetic diagnostics, knowledge about neuromuscular disorders, particularly CMDs, has increased dramatically. Thus, the incidence may be higher than originally thought.</p>\n </section>\n \n <section>\n \n <h3> Conclusion</h3>\n \n <p>This article reviews the recent achievements related to the clinical, diagnostic, pathogenic, and therapeutic aspects of CMDs.</p>\n </section>\n </div>","PeriodicalId":72232,"journal":{"name":"Annals of the Child Neurology Society","volume":"2 1","pages":"27-39"},"PeriodicalIF":0.0000,"publicationDate":"2023-12-20","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1002/cns3.20050","citationCount":"0","resultStr":"{\"title\":\"The congenital muscular dystrophies\",\"authors\":\"Haluk Topaloğlu, Bita Poorshiri\",\"doi\":\"10.1002/cns3.20050\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<div>\\n \\n \\n <section>\\n \\n <h3> Background</h3>\\n \\n <p>Congenital muscular dystrophies (CMDs) are genetically and clinically heterogeneous inherited conditions. Onset is typically within the first year of life. Most CMDs are autosomal recessive, except for de novo dominant mutations in <i>LMNA</i>-related muscular dystrophy and some collagen-6-associated disorders.</p>\\n </section>\\n \\n <section>\\n \\n <h3> Results</h3>\\n \\n <p>CMD is characterized by progressive muscular weakness, hypotonia, multiple contractures with a variable degree, spinal stiffness, delay in motor milestones acquisition, and histologically dystrophic lesions. Also, some forms of CMD may feature structural and myelination abnormalities on brain magnetic resonance imaging, intellectual impairment, and structural abnormalities of the eye. Muscle biopsy specimens exhibit a dystrophic pattern, but the appearance is quite variable depending on the different stages and severity of the disorder. The prevalence of CMD is estimated to be one in 100 000 people. Over the last few years, with advances in molecular genetic diagnostics, knowledge about neuromuscular disorders, particularly CMDs, has increased dramatically. Thus, the incidence may be higher than originally thought.</p>\\n </section>\\n \\n <section>\\n \\n <h3> Conclusion</h3>\\n \\n <p>This article reviews the recent achievements related to the clinical, diagnostic, pathogenic, and therapeutic aspects of CMDs.</p>\\n </section>\\n </div>\",\"PeriodicalId\":72232,\"journal\":{\"name\":\"Annals of the Child Neurology Society\",\"volume\":\"2 1\",\"pages\":\"27-39\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2023-12-20\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://onlinelibrary.wiley.com/doi/epdf/10.1002/cns3.20050\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Annals of the Child Neurology Society\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://onlinelibrary.wiley.com/doi/10.1002/cns3.20050\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"\",\"JCRName\":\"\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Annals of the Child Neurology Society","FirstCategoryId":"1085","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/cns3.20050","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"","JCRName":"","Score":null,"Total":0}
Congenital muscular dystrophies (CMDs) are genetically and clinically heterogeneous inherited conditions. Onset is typically within the first year of life. Most CMDs are autosomal recessive, except for de novo dominant mutations in LMNA-related muscular dystrophy and some collagen-6-associated disorders.
Results
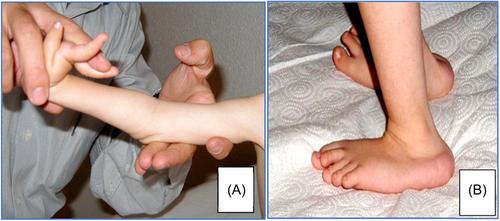
CMD is characterized by progressive muscular weakness, hypotonia, multiple contractures with a variable degree, spinal stiffness, delay in motor milestones acquisition, and histologically dystrophic lesions. Also, some forms of CMD may feature structural and myelination abnormalities on brain magnetic resonance imaging, intellectual impairment, and structural abnormalities of the eye. Muscle biopsy specimens exhibit a dystrophic pattern, but the appearance is quite variable depending on the different stages and severity of the disorder. The prevalence of CMD is estimated to be one in 100 000 people. Over the last few years, with advances in molecular genetic diagnostics, knowledge about neuromuscular disorders, particularly CMDs, has increased dramatically. Thus, the incidence may be higher than originally thought.
Conclusion
This article reviews the recent achievements related to the clinical, diagnostic, pathogenic, and therapeutic aspects of CMDs.



 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
